 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-8166 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
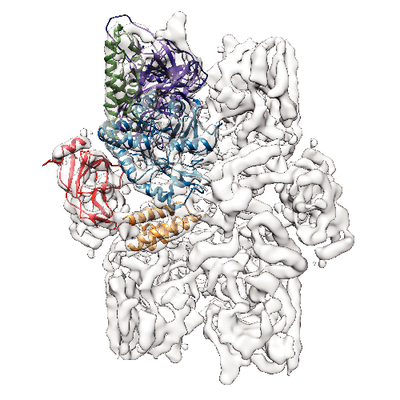
| Title | Structure of the S. cerevisiae alpha-mannosidase 1 | |||||||||
 Map data Map data | None | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | tetramer / cvt cargo / mannosidase / selective autophagy / hydrolase | |||||||||
| Function / homology |  Function and homology information Function and homology informationmannose catabolic process / Lysosomal oligosaccharide catabolism / Cvt complex / alpha-mannosidase / alpha-mannosidase activity / fungal-type vacuole membrane / oligosaccharide catabolic process / cellular response to nitrogen starvation / cellular response to glucose starvation / carbohydrate binding / metal ion binding Similarity search - Function | |||||||||
| Biological species |  | |||||||||
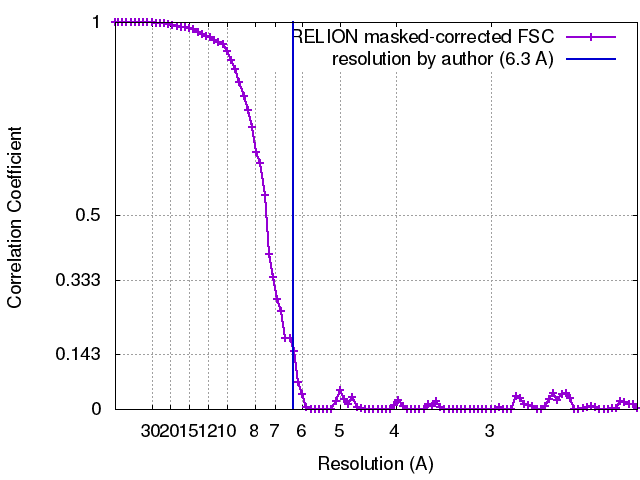
| Method | single particle reconstruction / cryo EM / Resolution: 6.3 Å | |||||||||
 Authors Authors | Schneider S / Kosinski J | |||||||||
 Citation Citation | Journal: EMBO Rep / Year: 2016 Title: Higher-order assemblies of oligomeric cargo receptor complexes form the membrane scaffold of the Cvt vesicle. Authors: Chiara Bertipaglia / Sarah Schneider / Arjen J Jakobi / Abul K Tarafder / Yury S Bykov / Andrea Picco / Wanda Kukulski / Jan Kosinski / Wim Jh Hagen / Arvind C Ravichandran / Matthias ...Authors: Chiara Bertipaglia / Sarah Schneider / Arjen J Jakobi / Abul K Tarafder / Yury S Bykov / Andrea Picco / Wanda Kukulski / Jan Kosinski / Wim Jh Hagen / Arvind C Ravichandran / Matthias Wilmanns / Marko Kaksonen / John Ag Briggs / Carsten Sachse /  Abstract: Selective autophagy is the mechanism by which large cargos are specifically sequestered for degradation. The structural details of cargo and receptor assembly giving rise to autophagic vesicles ...Selective autophagy is the mechanism by which large cargos are specifically sequestered for degradation. The structural details of cargo and receptor assembly giving rise to autophagic vesicles remain to be elucidated. We utilize the yeast cytoplasm-to-vacuole targeting (Cvt) pathway, a prototype of selective autophagy, together with a multi-scale analysis approach to study the molecular structure of Cvt vesicles. We report the oligomeric nature of the major Cvt cargo Ape1 with a combined 2.8 Å X-ray and negative stain EM structure, as well as the secondary cargo Ams1 with a 6.3 Å cryo-EM structure. We show that the major dodecameric cargo prApe1 exhibits a tendency to form higher-order chain structures that are broken upon interaction with the receptor Atg19 in vitro The stoichiometry of these cargo-receptor complexes is key to maintaining the size of the Cvt aggregate in vivo Using correlative light and electron microscopy, we further visualize key stages of Cvt vesicle biogenesis. Our findings suggest that Atg19 interaction limits Ape1 aggregate size while serving as a vehicle for vacuolar delivery of tetrameric Ams1. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_8166.map.gz | 6.4 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-8166-v30.xmlemd-8166.xml | 16.4 KB 16.4 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_8166_fsc.xml | 8.6 KB | Display | FSC data file |
| Images |  emd_8166.png emd_8166.png | 427.6 KB | ||
| Filedesc metadata | emd-8166.cif.gz | 7.2 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-8166ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8166 http://ftp.pdbj.org/pub/emdb/structures/EMD-8166ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8166 | HTTPS FTP |
-Related structure data
| Related structure data |  5jm0MC  8167C  5jm6C  5jm9C M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |








-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |



-Map
| File | Download / File: emd_8166.map.gz / Format: CCP4 / Size: 59.6 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | None | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.08 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
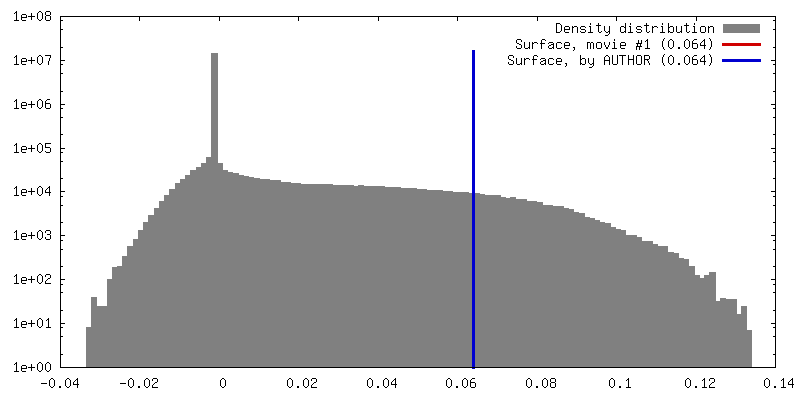
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)



















-Supplemental data
- Sample components
Sample components
-Entire : Alpha-mannosidase 1
| Entire | Name: Alpha-mannosidase 1 |
|---|---|
| Components |
|
-Supramolecule #1: Alpha-mannosidase 1
| Supramolecule | Name: Alpha-mannosidase 1 / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all / Details: His-tagged |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 500 KDa |
-Macromolecule #1: Alpha-mannosidase,Alpha-mannosidase,Alpha-mannosidase
| Macromolecule | Name: Alpha-mannosidase,Alpha-mannosidase,Alpha-mannosidase / type: protein_or_peptide / ID: 1 Details: The coordinate model contains a poly-alanine stretch that corresponds to residues 17-27 in the template sequence. Number of copies: 1 / Enantiomer: LEVO / EC number: alpha-mannosidase |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 125.764156 KDa |
| Recombinant expression | Organism:  Komagataella pastoris (fungus) Komagataella pastoris (fungus) |
| Sequence | String: (UNK)(UNK)(UNK)(UNK)(UNK)(UNK)(UNK)(UNK)(UNK)(UNK) (UNK)(UNK)(UNK)MSSEDII YDPQFKPV Q GIYENRLRQF IDTGGDYHDL NLPKFYDKKR ISLDHDHVKV WWYQVSFERG SSPVSPDKRP SWKSIIERDK KGELEFREA NINQPFGPSW ...String: (UNK)(UNK)(UNK)(UNK)(UNK)(UNK)(UNK)(UNK)(UNK)(UNK) (UNK)(UNK)(UNK)MSSEDII YDPQFKPV Q GIYENRLRQF IDTGGDYHDL NLPKFYDKKR ISLDHDHVKV WWYQVSFERG SSPVSPDKRP SWKSIIERDK KGELEFREA NINQPFGPSW STTWFKVKIS LPEDWVKSNE QLLFQWDCSN EGIVIDPKTL IPVTAFSGGE RTEYVLPKTS DGKHFFYIEA GNNGMFGCG AGSTINPPDD NRFFHLRKAD IVWPDLDARA LYIDFWMLGD AARELPGDSW QKHQARQLGN AVMNLFDPND R SSVRKCRE LLQREYFDSF LESSKVYEQG ESQVLTNVYG IGNCHIDTAW LWPFAETRRK IVRSWSSQCT LMDRFPEYKF VA SQAQQFK WLLEDHPEFF NKVLIPKIQQ SQFFAVGGTW VENDTNIPSG ESLARQFFFG QRFFLKHFGL KSKIFWLPDT FGY SSQMPQ LCRLSGIDKF LTQKLSWNNI NSFPHSTFNW AGIDGSQLLT HMPPGNTYTA DSHFGDVLRT AKQNKTPEYY GSGL MLYGK GDGGGGPTEE MLQKMRRIRS MNNRNGNVIP KLQVGITVDE FYDDILKRTN QGHDLPTWSG ELYFEFHRGT YTSQA QTKK LMRLSEIKLH DLEWIAAKTS VLYPDSYKYP SKQINELWEN VLLCQFHDVL PGSCIEMVYK YEAVPMLHNV VKECTS LID KTVQFLQSQS KADLVEMRTL TWSKPEKVSE ECSLNGSYTS SVTGYDDYIV LANGKLKVII CKKTGVITSI TDETLGV EY LDTEHGRNKL GANQFVIYDD KPLGWQAWDT ELYSVNQYKY VTKPKKVQVS CNTKEKCAVE VIFQISEKCK IKSVISLN A TAVTDAKLSK VDISTTVENW DARNKFLKVE FPVNIRNDFA SYETQFGITK RPTHYNTSWD VAKFEVCHHK FADYSEYSK GVSILNDCKY GFSTHGNLMR LSLLRSPKAP DAHADMGTHE IKYAIYPHRG ALSSDTVKLA HEFNYCFKYK LPKDIGMNFD DIISISGDE NVILSNIKRG EDDSAVKSNY SLNPRDEQSI VVRVYESLGG ESFASLNTTL NLKRIEKVDN LEMKVYKSLT A TRDESNHA INRIPIKLRP FEIASFRLYF UniProtKB: Alpha-mannosidase |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.4 mg/mL | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.5 Component:
| ||||||||||||
| Grid | Model: Quantifoil R2/2 / Material: COPPER / Mesh: 300 / Support film - Material: CARBON / Support film - topology: HOLEY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 60 sec. / Pretreatment - Atmosphere: AIR / Pretreatment - Pressure: 4.5 kPa | ||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 95 % / Chamber temperature: 293 K / Instrument: FEI VITROBOT MARK III Details: 2.5 ul of sample was applied, offset -3, 9s blotting time. | ||||||||||||
| Details | single particles alongside chains of tetramers |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: FEI FALCON II (4k x 4k) / Detector mode: INTEGRATING / Digitization - Frames/image: 1-7 / Number grids imaged: 1 / Number real images: 1735 / Average exposure time: 1.6 sec. / Average electron dose: 58.0 e/Å2 Details: The first six frames comprised each 7 e/A2 and the final frame consequently received 16 e/A2 dose. Relion's particle polishing procedure was used. |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 50.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 5.0 µm / Nominal defocus min: 1.0 µm / Nominal magnification: 75000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing
-Atomic model buiding 1
| Details | 1. Fitting of a homology model based on PDB 2wyh combined with a homology model based on PDB 2xbz and 3cmg for the C- (residues 287-1083) and N-terminal part (residues 45-203) respectively 2. ab initio modelling for a three-helix bundle of the N-terminal part (residues 209-286) 3. creating an ideal poly-alanine helix for residues 17-27 |
|---|---|
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT / Overall B value: 80 / Target criteria: Cross-correlation coefficient |
| Output model | PDB-5jm0: |
