 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
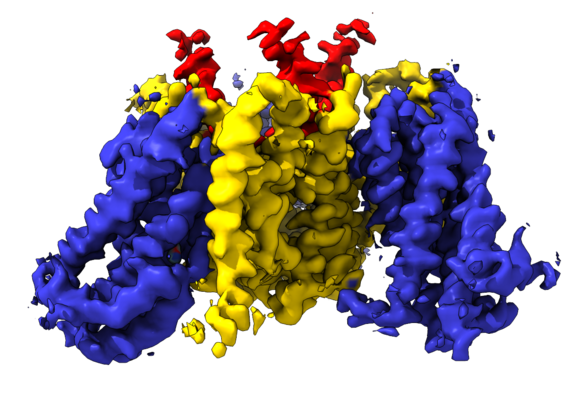
Yorodumi- EMDB-4386: cryo-EM structure of the human neutral amino acid transporter ASCT2 -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-4386 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | cryo-EM structure of the human neutral amino acid transporter ASCT2 | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | neutral amino acid transporter / MEMBRANE PROTEIN | |||||||||
| Function / homology |  Function and homology information Function and homology informationL-glutamine secretion / L-glutamine import across plasma membrane / L-glutamine transmembrane transporter activity / L-serine transmembrane transporter activity / L-glutamine transport / ligand-gated channel activity / L-aspartate transmembrane transporter activity / neutral amino acid transport / L-aspartate import across plasma membrane / neutral L-amino acid transmembrane transporter activity ...L-glutamine secretion / L-glutamine import across plasma membrane / L-glutamine transmembrane transporter activity / L-serine transmembrane transporter activity / L-glutamine transport / ligand-gated channel activity / L-aspartate transmembrane transporter activity / neutral amino acid transport / L-aspartate import across plasma membrane / neutral L-amino acid transmembrane transporter activity / symporter activity / Amino acid transport across the plasma membrane / amino acid transmembrane transporter activity / antiporter activity / RHOJ GTPase cycle / protein homotrimerization / RHOQ GTPase cycle / amino acid transport / RHOH GTPase cycle / RAC3 GTPase cycle / transport across blood-brain barrier / RAC1 GTPase cycle / basal plasma membrane / erythrocyte differentiation / centriolar satellite / melanosome / virus receptor activity / signaling receptor activity / ciliary basal body / extracellular exosome / membrane / metal ion binding / plasma membrane Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.85 Å | |||||||||
 Authors Authors | Garaeva AA / Oostergetel GT | |||||||||
| Funding support |  Netherlands, 2 items Netherlands, 2 items
| |||||||||
 Citation Citation | Journal: Nat Struct Mol Biol / Year: 2018 Title: Cryo-EM structure of the human neutral amino acid transporter ASCT2. Authors: Alisa A Garaeva / Gert T Oostergetel / Cornelius Gati / Albert Guskov / Cristina Paulino / Dirk J Slotboom /  Abstract: Human ASCT2 belongs to the SLC1 family of secondary transporters and is specific for the transport of small neutral amino acids. ASCT2 is upregulated in cancer cells and serves as the receptor for ...Human ASCT2 belongs to the SLC1 family of secondary transporters and is specific for the transport of small neutral amino acids. ASCT2 is upregulated in cancer cells and serves as the receptor for many retroviruses; hence, it has importance as a potential drug target. Here we used single-particle cryo-EM to determine a structure of the functional and unmodified human ASCT2 at 3.85-Å resolution. ASCT2 forms a homotrimeric complex in which each subunit contains a transport and a scaffold domain. Prominent extracellular extensions on the scaffold domain form the predicted docking site for retroviruses. Relative to structures of other SLC1 members, ASCT2 is in the most extreme inward-oriented state, with the transport domain largely detached from the central scaffold domain on the cytoplasmic side. This domain detachment may be required for substrate binding and release on the intracellular side of the membrane. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_4386.map.gz | 48.8 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-4386-v30.xmlemd-4386.xml | 24.2 KB 24.2 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_4386_fsc.xml | 8.6 KB | Display | FSC data file |
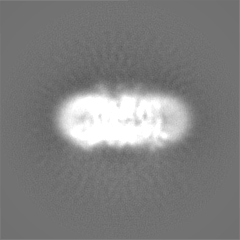
| Images |  emd_4386.png emd_4386.png | 223.3 KB | ||
| Masks | emd_4386_msk_1.map | 52.7 MB | Mask map | |
| Filedesc metadata | emd-4386.cif.gz | 7.9 KB | ||
| Others | emd_4386_additional.map.gzemd_4386_half_map_1.map.gzemd_4386_half_map_2.map.gz | 49.1 MB 40.6 MB 40.6 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-4386ftp://ftp.pdbj.org/pub/emdb/structures/EMD-4386 http://ftp.pdbj.org/pub/emdb/structures/EMD-4386ftp://ftp.pdbj.org/pub/emdb/structures/EMD-4386 | HTTPS FTP |
-Related structure data
| Related structure data |  6gctMC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |








-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |






-Map
| File | Download / File: emd_4386.map.gz / Format: CCP4 / Size: 52.7 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.012 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Mask #1
| File | emd_4386_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |






-Additional map: #1
| File | emd_4386_additional.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: half-map 1 used for post processing step and...
| File | emd_4386_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | half-map 1 used for post processing step and FSC resolution calculation. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |






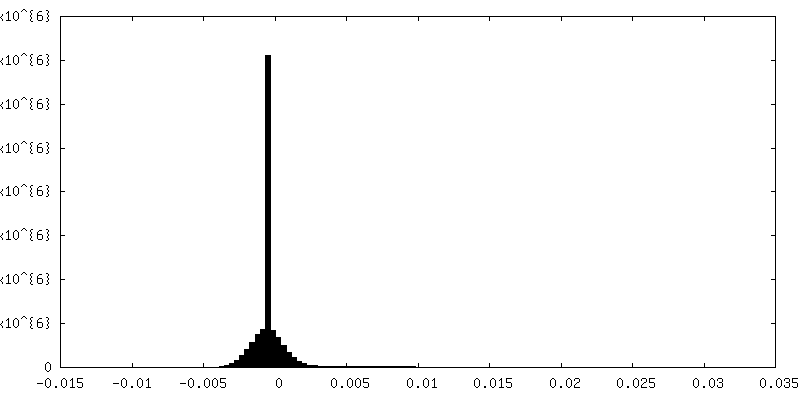

-Half map: half-map 2 used for post processing step and...
| File | emd_4386_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | half-map 2 used for post processing step and FSC resolution calculation. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |







- Sample components
Sample components
-Entire : human ASCT2 SLC1A5
| Entire | Name: human ASCT2 SLC1A5 |
|---|---|
| Components |
|
-Supramolecule #1: human ASCT2 SLC1A5
| Supramolecule | Name: human ASCT2 SLC1A5 / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 / Details: human ASCT2 SLC1A5 |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Molecular weight | Theoretical: 172 KDa |
-Macromolecule #1: Neutral amino acid transporter B(0)
| Macromolecule | Name: Neutral amino acid transporter B(0) / type: protein_or_peptide / ID: 1 / Number of copies: 3 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Molecular weight | Theoretical: 56.638902 KDa |
| Recombinant expression | Organism:  Komagataella pastoris (fungus) Komagataella pastoris (fungus) |
| Sequence | String: MVADPPRDSK GLAAAEPTAN GGLALASIED QGAAAGGYCG SRDQVRRCLR ANLLVLLTVV AVVAGVALGL GVSGAGGALA LGPERLSAF VFPGELLLRL LRMIILPLVV CSLIGGAASL DPGALGRLGA WALLFFLVTT LLASALGVGL ALALQPGAAS A AINASVGA ...String: MVADPPRDSK GLAAAEPTAN GGLALASIED QGAAAGGYCG SRDQVRRCLR ANLLVLLTVV AVVAGVALGL GVSGAGGALA LGPERLSAF VFPGELLLRL LRMIILPLVV CSLIGGAASL DPGALGRLGA WALLFFLVTT LLASALGVGL ALALQPGAAS A AINASVGA AGSAENAPSK EVLDSFLDLA RNIFPSNLVS AAFRSYSTTY EERNITGTRV KVPVGQEVEG MNILGLVVFA IV FGVALRK LGPEGELLIR FFNSFNEATM VLVSWIMWYA PVGIMFLVAG KIVEMEDVGL LFARLGKYIL CCLLGHAIHG LLV LPLIYF LFTRKNPYRF LWGIVTPLAT AFGTSSSSAT LPLMMKCVEE NNGVAKHISR FILPIGATVN MDGAALFQCV AAVF IAQLS QQSLDFVKII TILVTATASS VGAAGIPAGG VLTLAIILEA VNLPVDHISL ILAVDWLVDR SCTVLNVEGD ALGAG LLQN YVDRTESRST EPELIQVKSE LPLDPLPVPT EEGNPLLKHY RGPAGDATVA SEKESVM UniProtKB: Neutral amino acid transporter B(0) |
-Macromolecule #2: GLUTAMINE
| Macromolecule | Name: GLUTAMINE / type: ligand / ID: 2 / Number of copies: 3 / Formula: GLN |
|---|---|
| Molecular weight | Theoretical: 146.144 Da |
| Chemical component information |  ChemComp-GLN: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 2.5 mg/mL |
|---|---|
| Buffer | pH: 7 Details: 20mM Tris-HCl pH 7.4 300mM NaCl 1mM L-glutamine 0.05% DDM 0.005% CHS |
| Grid | Model: Quantifoil, UltrAuFoil, R1.2/1.3 / Material: GOLD / Mesh: 200 / Support film - Material: CARBON / Support film - topology: HOLEY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 20 sec. |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 278 K / Instrument: FEI VITROBOT MARK II |
- Electron microscopy
Electron microscopy
| Microscope | FEI TALOS ARCTICA |
|---|---|
| Temperature | Min: 70.0 K / Max: 90.0 K |
| Specialist optics | Energy filter - Name: GIF Quantum LS / Energy filter - Lower energy threshold: 0 eV / Energy filter - Upper energy threshold: 20 eV |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Digitization - Frames/image: 1-60 / Average exposure time: 9.0 sec. / Average electron dose: 0.87 e/Å2 Details: Freshly purified protein was concentrated using Vivaspin concentrating devices with a molecular weight cutoff of 100kDa to 2-2.5 mg ml-1. 2.8 ul were applied on holey-carbon cryo-EM grids ...Details: Freshly purified protein was concentrated using Vivaspin concentrating devices with a molecular weight cutoff of 100kDa to 2-2.5 mg ml-1. 2.8 ul were applied on holey-carbon cryo-EM grids (Quantifoil Au R1.2-1.3, 200 and 300 mesh), which were prior glow-discharged at 5 mA for 20 s. Grids were blotted for 3-5 s in a Vitrobot (Mark 3, Thermo Fisher) at 20C temperature and 100% humidity, subsequently plunge-frozen in liquid ethane and stored in liquid nitrogen. Cryo-EM data were collected on a 200 keV Talos Arctica microscope (Thermo Fisher) using a post-column energy filter (Gatan) in zero-loss mode, using a 20 eV slit, a 100 um objective aperture, in an automated fashion using EPU software (Thermo Fisher) on a K2 summit detector (Gatan) in counting mode. Cryo-EM images were acquired at a pixel size of 1.012A (calibrated magnification of 49,407x), a defocus range from -0.4 to 2.5 um, an exposure time of 9 sec and a sub-frame exposure time of 150 ms (60 frames), and a total electron dose on the specimen level of about 52 electrons per A2. Best regions on the grid were screened with a self-written script to calculate the ice thickness and data quality was monitored on the fly using the software FOCUS |
| Electron beam | Acceleration voltage: 200 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 100.0 µm / Calibrated defocus max: 0.0025 µm / Calibrated defocus min: 0.0004 µm / Calibrated magnification: 49407 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 0.0025 µm / Nominal defocus min: 0.0004 µm / Nominal magnification: 49407 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Talos Arctica / Image courtesy: FEI Company |
+Image processing


-Atomic model buiding 1
| Refinement | Space: REAL / Protocol: AB INITIO MODEL |
|---|---|
| Output model | PDB-6gct: |
