 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: EMDB / ID: EMD-4094 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
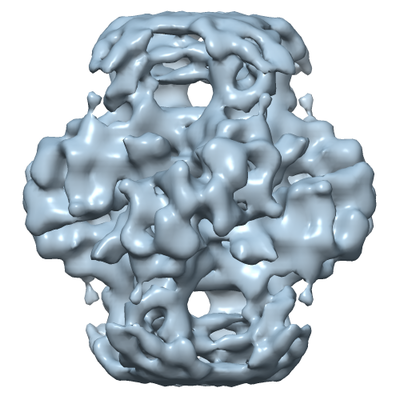
| タイトル | Structure of a designed Isoaspartyl Dipeptidase filament. | |||||||||
 マップデータ マップデータ | ||||||||||
 試料 試料 |
| |||||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報hydrolase activity, acting on carbon-nitrogen (but not peptide) bonds / 加水分解酵素; プロテアーゼ; ペプチド結合加水分解酵素; オメガペプチターゼ / beta-aspartyl-peptidase activity / metallopeptidase activity / proteolysis / zinc ion binding / identical protein binding / cytosol / cytoplasm 類似検索 - 分子機能 | |||||||||
| 生物種 |  | |||||||||
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 10.5 Å | |||||||||
 データ登録者 データ登録者 | Garcia-Seisdedos H / Empereur-Mot C / Elad N / Levy DE | |||||||||
 引用 引用 | ジャーナル: Nature / 年: 2017 タイトル: Proteins evolve on the edge of supramolecular self-assembly. 著者: Hector Garcia-Seisdedos / Charly Empereur-Mot / Nadav Elad / Emmanuel D Levy /  要旨: The self-association of proteins into symmetric complexes is ubiquitous in all kingdoms of life. Symmetric complexes possess unique geometric and functional properties, but their internal symmetry ...The self-association of proteins into symmetric complexes is ubiquitous in all kingdoms of life. Symmetric complexes possess unique geometric and functional properties, but their internal symmetry can pose a risk. In sickle-cell disease, the symmetry of haemoglobin exacerbates the effect of a mutation, triggering assembly into harmful fibrils. Here we examine the universality of this mechanism and its relation to protein structure geometry. We introduced point mutations solely designed to increase surface hydrophobicity among 12 distinct symmetric complexes from Escherichia coli. Notably, all responded by forming supramolecular assemblies in vitro, as well as in vivo upon heterologous expression in Saccharomyces cerevisiae. Remarkably, in four cases, micrometre-long fibrils formed in vivo in response to a single point mutation. Biophysical measurements and electron microscopy revealed that mutants self-assembled in their folded states and so were not amyloid-like. Structural examination of 73 mutants identified supramolecular assembly hot spots predictable by geometry. A subsequent structural analysis of 7,471 symmetric complexes showed that geometric hot spots were buffered chemically by hydrophilic residues, suggesting a mechanism preventing mis-assembly of these regions. Thus, point mutations can frequently trigger folded proteins to self-assemble into higher-order structures. This potential is counterbalanced by negative selection and can be exploited to design nanomaterials in living cells. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| ムービー |
ムービービューア |
|---|---|
| 構造ビューア | EMマップ: SurfViewMolmilJmol/JSmol |
| 添付画像 |
- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_4094.map.gz | 7.3 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-4094-v30.xmlemd-4094.xml | 12.2 KB 12.2 KB | 表示 表示 | EMDBヘッダ |
| 画像 |  emd_4094.png emd_4094.png | 155.2 KB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-4094ftp://ftp.pdbj.org/pub/emdb/structures/EMD-4094 http://ftp.pdbj.org/pub/emdb/structures/EMD-4094ftp://ftp.pdbj.org/pub/emdb/structures/EMD-4094 | HTTPS FTP |
-検証レポート
| 文書・要旨 | emd_4094_validation.pdf.gz | 229.9 KB | 表示 | EMDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | emd_4094_full_validation.pdf.gz | 229.1 KB | 表示 | |
| XML形式データ | emd_4094_validation.xml.gz | 5.2 KB | 表示 | |
| アーカイブディレクトリ | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-4094ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-4094 | HTTPS FTP |
-関連構造データ






-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| 「今月の分子」の関連する項目 |


-マップ
| ファイル | ダウンロード / ファイル: emd_4094.map.gz / 形式: CCP4 / 大きさ: 8 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 投影像・断面図 | 画像のコントロール
画像は Spider により作成 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 2.11 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 密度 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
CCP4マップ ヘッダ情報:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-添付データ
- 試料の構成要素
試料の構成要素
-全体 : Fiber assembly of isoaspartyl dipeptidase E239Y mutant
| 全体 | 名称: Fiber assembly of isoaspartyl dipeptidase E239Y mutant |
|---|---|
| 要素 |
|
-超分子 #1: Fiber assembly of isoaspartyl dipeptidase E239Y mutant
| 超分子 | 名称: Fiber assembly of isoaspartyl dipeptidase E239Y mutant タイプ: complex / ID: 1 / 親要素: 0 / 含まれる分子: all 詳細: E239Y point mutant induce fiber formation of the isoaspartyl dipeptidase octamers |
|---|---|
| 由来(天然) | 生物種: |
| 組換発現 | 生物種: 組換プラスミド: pET-30 a (+) |
-分子 #1: Isoaspartyl Dipeptidase
| 分子 | 名称: Isoaspartyl Dipeptidase / タイプ: protein_or_peptide / ID: 1 / 光学異性体: LEVO / EC番号: beta-aspartyl-peptidase |
|---|---|
| 由来(天然) | 生物種: |
| 組換発現 | 生物種: |
| 配列 | 文字列: HHHHHHLVPR GIDYTAAGFT LLQGAHLYA PEDRGICDVL V ANGKIIAV ASNIPSDIVP NC TVVDLSG QILCPGFIDQ HVH LIGGGG EAGPTTRTPE VALS RLTEA GVTSVVGLLG TDSIS RHPE SLLAKTRALN EEGISA WML TGAYHVPSRT ITGSVEK DV ...文字列: HHHHHHLVPR GIDYTAAGFT LLQGAHLYA PEDRGICDVL V ANGKIIAV ASNIPSDIVP NC TVVDLSG QILCPGFIDQ HVH LIGGGG EAGPTTRTPE VALS RLTEA GVTSVVGLLG TDSIS RHPE SLLAKTRALN EEGISA WML TGAYHVPSRT ITGSVEK DV AIIDRVIGVK CAISDHRS A APDVYHLANM AAESRVGGL LGGKPGVTVF HMGDSKKALQ PIYDLLENC DVPISKLLPT H VNRNVPLF YQALEFARKG GT IDITSSI DEPVAPAEGI ARA VQAGIP LARVTLSSDG NGSQ PFFDD EGNLTHIGVA GFETL LETV QVLVKDYDFS ISDALR PLT SSVAGFLNLT GKGEILP GN DADLLVMTPE LRIEQVYA R GKLMVKDGKA CVKGTFETA |
-実験情報
-構造解析
| 手法 | クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | filament |
-試料調製
| 濃度 | 0.2 mg/mL | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 緩衝液 | pH: 7.5 構成要素:
| |||||||||
| グリッド | モデル: Quantifoil / 材質: COPPER / メッシュ: 300 / 支持フィルム - 材質: CARBON / 支持フィルム - トポロジー: CONTINUOUS / 前処理 - タイプ: GLOW DISCHARGE / 前処理 - 雰囲気: AIR | |||||||||
| 凍結 | 凍結剤: ETHANE / チャンバー内湿度: 95 % / チャンバー内温度: 297 K / 装置: LEICA EM GP |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TECNAI F20 |
|---|---|
| 撮影 | フィルム・検出器のモデル: GATAN K2 SUMMIT (4k x 4k) 平均電子線量: 32.0 e/Å2 |
| 電子線 | 加速電圧: 200 kV / 電子線源:  FIELD EMISSION GUN FIELD EMISSION GUN |
| 電子光学系 | C2レンズ絞り径: 30.0 µm / 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELD |
| 実験機器 |  モデル: Tecnai F20 / 画像提供: FEI Company |
+画像解析

-原子モデル構築 1
| 精密化 | プロトコル: RIGID BODY FIT |
|---|---|
| 得られたモデル |  PDB-5lp3: |
