 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-35983: Cryo-EM structure of Mycobacterium tuberculosis ATP synthase Fo i... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM structure of Mycobacterium tuberculosis ATP synthase Fo in the apo-form | ||||||||||||
 Map data Map data | |||||||||||||
 Sample Sample |
| ||||||||||||
 Keywords Keywords | ATP synthase / Mycobacterium tuberculosis / cryo-EM / MEMBRANE PROTEIN | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationproton-transporting ATP synthase complex, coupling factor F(o) / proton-transporting ATP synthase activity, rotational mechanism / hydrolase activity / lipid binding / plasma membrane Similarity search - Function | ||||||||||||
| Biological species |   Mycobacterium tuberculosis (bacteria) Mycobacterium tuberculosis (bacteria) | ||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.15 Å | ||||||||||||
 Authors Authors | Zhang Y / Lai Y / Liu F / Rao Z / Gong H | ||||||||||||
| Funding support |  China, 3 items China, 3 items
| ||||||||||||
 Citation Citation | Journal: Nature / Year: 2024 Title: Inhibition of M. tuberculosis and human ATP synthase by BDQ and TBAJ-587. Authors: Yuying Zhang / Yuezheng Lai / Shan Zhou / Ting Ran / Yue Zhang / Ziqing Zhao / Ziyan Feng / Long Yu / Jinxu Xu / Kun Shi / Jianyun Wang / Yu Pang / Liang Li / Hongming Chen / Luke W Guddat / ...Authors: Yuying Zhang / Yuezheng Lai / Shan Zhou / Ting Ran / Yue Zhang / Ziqing Zhao / Ziyan Feng / Long Yu / Jinxu Xu / Kun Shi / Jianyun Wang / Yu Pang / Liang Li / Hongming Chen / Luke W Guddat / Yan Gao / Fengjiang Liu / Zihe Rao / Hongri Gong /  Abstract: Bedaquiline (BDQ), a first-in-class diarylquinoline anti-tuberculosis drug, and its analogue, TBAJ-587, prevent the growth and proliferation of Mycobacterium tuberculosis by inhibiting ATP synthase. ...Bedaquiline (BDQ), a first-in-class diarylquinoline anti-tuberculosis drug, and its analogue, TBAJ-587, prevent the growth and proliferation of Mycobacterium tuberculosis by inhibiting ATP synthase. However, BDQ also inhibits human ATP synthase. At present, how these compounds interact with either M. tuberculosis ATP synthase or human ATP synthase is unclear. Here we present cryogenic electron microscopy structures of M. tuberculosis ATP synthase with and without BDQ and TBAJ-587 bound, and human ATP synthase bound to BDQ. The two inhibitors interact with subunit a and the c-ring at the leading site, c-only sites and lagging site in M. tuberculosis ATP synthase, showing that BDQ and TBAJ-587 have similar modes of action. The quinolinyl and dimethylamino units of the compounds make extensive contacts with the protein. The structure of human ATP synthase in complex with BDQ reveals that the BDQ-binding site is similar to that observed for the leading site in M. tuberculosis ATP synthase, and that the quinolinyl unit also interacts extensively with the human enzyme. This study will improve researchers' understanding of the similarities and differences between human ATP synthase and M. tuberculosis ATP synthase in terms of the mode of BDQ binding, and will allow the rational design of novel diarylquinolines as anti-tuberculosis drugs. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_35983.map.gz | 483.5 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-35983-v30.xmlemd-35983.xml | 16.1 KB 16.1 KB | Display Display | EMDB header |
| Images |  emd_35983.png emd_35983.png | 35.5 KB | ||
| Filedesc metadata | emd-35983.cif.gz | 5.7 KB | ||
| Others | emd_35983_half_map_1.map.gzemd_35983_half_map_2.map.gz | 475.7 MB 475.7 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-35983ftp://ftp.pdbj.org/pub/emdb/structures/EMD-35983 http://ftp.pdbj.org/pub/emdb/structures/EMD-35983ftp://ftp.pdbj.org/pub/emdb/structures/EMD-35983 | HTTPS FTP |
-Validation report
| Summary document | emd_35983_validation.pdf.gz | 1.2 MB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_35983_full_validation.pdf.gz | 1.2 MB | Display | |
| Data in XML | emd_35983_validation.xml.gz | 19 KB | Display | |
| Data in CIF | emd_35983_validation.cif.gz | 22.5 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-35983ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-35983 | HTTPS FTP |
-Related structure data
| Related structure data |  8j58MC  8j0sC  8j0tC 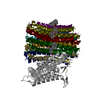 8j57C  8jr0C  8jr1C  8khfC  8ki3C M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |















-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |


-Map
| File | Download / File: emd_35983.map.gz / Format: CCP4 / Size: 512 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
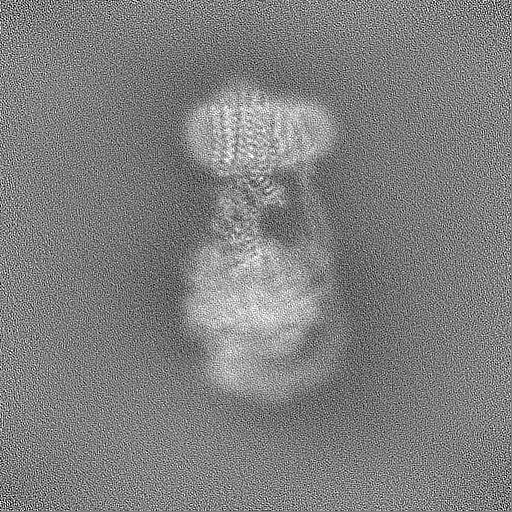
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.73 Å | ||||||||||||||||||||||||||||||||||||
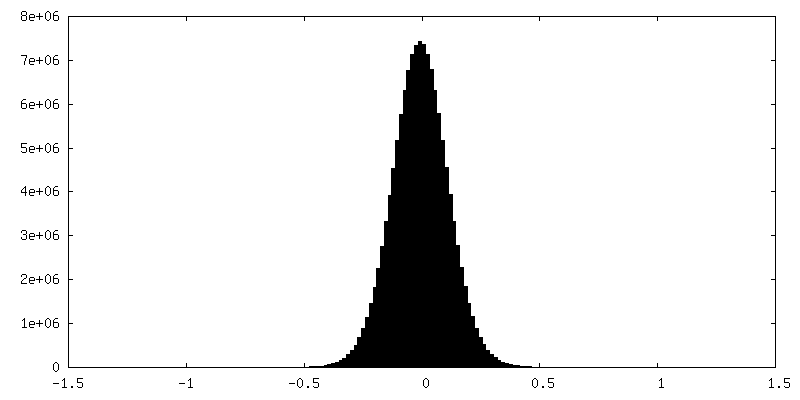
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Half map: #2
| File | emd_35983_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
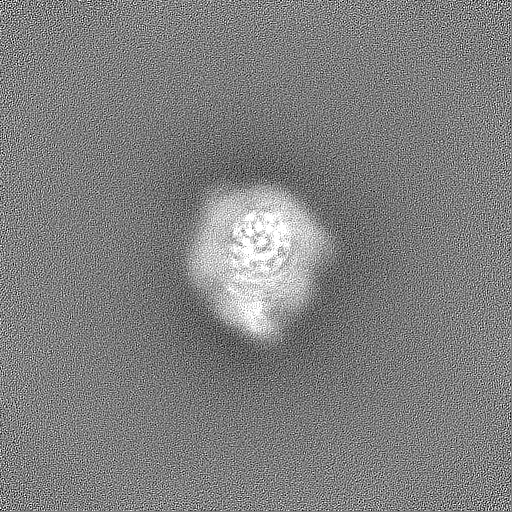
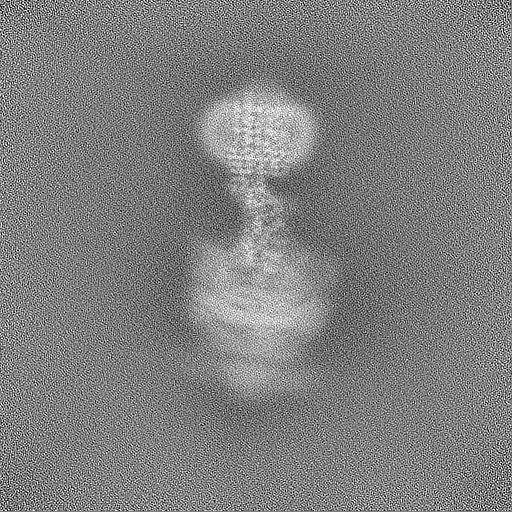
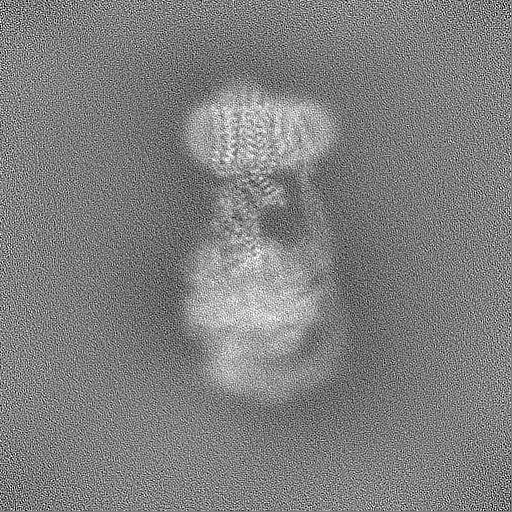
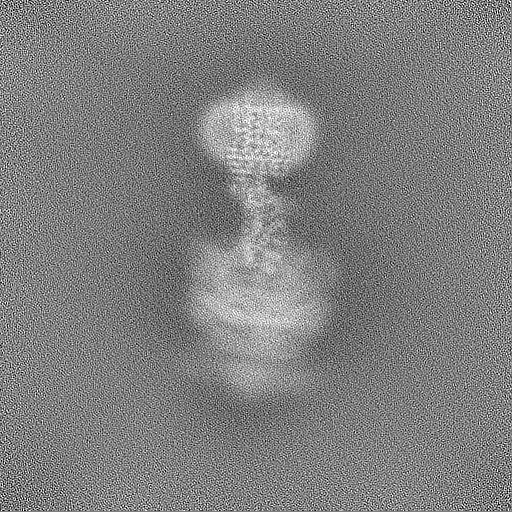
| Projections & Slices |
| ||||||||||||
| Density Histograms |


-Half map: #1
| File | emd_35983_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |




- Sample components
Sample components
-Entire : Mycobacterium tuberculosis ATP synthase Fo in the apo-form
| Entire | Name: Mycobacterium tuberculosis ATP synthase Fo in the apo-form |
|---|---|
| Components |
|
-Supramolecule #1: Mycobacterium tuberculosis ATP synthase Fo in the apo-form
| Supramolecule | Name: Mycobacterium tuberculosis ATP synthase Fo in the apo-form type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Mycobacterium tuberculosis (bacteria) |
-Macromolecule #1: ATP synthase subunit c
| Macromolecule | Name: ATP synthase subunit c / type: protein_or_peptide / ID: 1 / Number of copies: 9 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Mycobacterium tuberculosis (bacteria) |
| Molecular weight | Theoretical: 8.058423 KDa |
| Recombinant expression | Organism: Mycolicibacterium smegmatis (bacteria) |
| Sequence | String: MDPTIAAGAL IGGGLIMAGG AIGAGIGDGV AGNALISGVA RQPEAQGRLF TPFFITVGLV EAAYFINLAF MALFVFATPV K UniProtKB: ATP synthase subunit c |
-Macromolecule #2: ATP synthase subunit a
| Macromolecule | Name: ATP synthase subunit a / type: protein_or_peptide / ID: 2 / Number of copies: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Mycobacterium tuberculosis (bacteria) |
| Molecular weight | Theoretical: 27.488436 KDa |
| Recombinant expression | Organism: Mycolicibacterium smegmatis (bacteria) |
| Sequence | String: MTETILAAQI EVGEHHTATW LGMTVNTDTV LSTAIAGLIV IALAFYLRAK VTSTDVPGGV QLFFEAITIQ MRNQVESAIG MRIAPFVLP LAVTIFVFIL ISNWLAVLPV QYTDKHGHTT ELLKSAAADI NYVLALALFV FVCYHTAGIW RRGIVGHPIK L LKGHVTLL ...String: MTETILAAQI EVGEHHTATW LGMTVNTDTV LSTAIAGLIV IALAFYLRAK VTSTDVPGGV QLFFEAITIQ MRNQVESAIG MRIAPFVLP LAVTIFVFIL ISNWLAVLPV QYTDKHGHTT ELLKSAAADI NYVLALALFV FVCYHTAGIW RRGIVGHPIK L LKGHVTLL APINLVEEVA KPISLSLRLF GNIFAGGILV ALIALFPPYI MWAPNAIWKA FDLFVGAIQA FIFALLTILY FS QAMELEE EHH UniProtKB: ATP synthase subunit a |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 7.4 |
|---|---|
| Grid | Model: Quantifoil R1.2/1.3 / Material: COPPER |
| Vitrification | Cryogen name: ETHANE |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Specialist optics | Energy filter - Name: TFS Selectris X / Energy filter - Slit width: 10 eV |
| Image recording | Film or detector model: FEI FALCON IV (4k x 4k) / Average electron dose: 50.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Nominal defocus max: 2.4 µm / Nominal defocus min: 1.2 µm |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Startup model | Type of model: OTHER / Details: AlphaFold |
|---|---|
| Final reconstruction | Resolution.type: BY AUTHOR / Resolution: 3.15 Å / Resolution method: FSC 0.143 CUT-OFF / Number images used: 66593 |
| Initial angle assignment | Type: MAXIMUM LIKELIHOOD |
| Final angle assignment | Type: MAXIMUM LIKELIHOOD |
