 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-3296 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
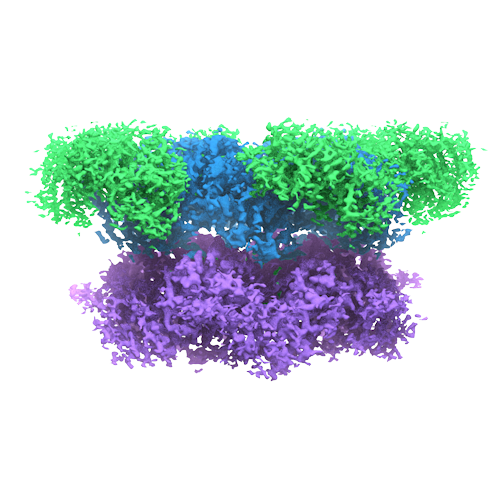
| Title | Cryo-EM structure of human p97 bound to ADP | |||||||||
 Map data Map data | Reconstruction of human p97 bound to ADP. | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | cryo-electron microscopy / single-particle / p97 / AAA ATPase | |||||||||
| Function / homology |  Function and homology information Function and homology informationprotein binding / flavin adenine dinucleotide catabolic process / VCP-NSFL1C complex / endoplasmic reticulum stress-induced pre-emptive quality control / endosome to lysosome transport via multivesicular body sorting pathway / BAT3 complex binding / cellular response to arsenite ion / cytoplasmic ubiquitin ligase complex / protein-DNA covalent cross-linking repair / Derlin-1 retrotranslocation complex ...protein binding / flavin adenine dinucleotide catabolic process / VCP-NSFL1C complex / endoplasmic reticulum stress-induced pre-emptive quality control / endosome to lysosome transport via multivesicular body sorting pathway / BAT3 complex binding / cellular response to arsenite ion / cytoplasmic ubiquitin ligase complex / protein-DNA covalent cross-linking repair / Derlin-1 retrotranslocation complex / positive regulation of protein K63-linked deubiquitination / ATPase complex / deubiquitinase activator activity / positive regulation of oxidative phosphorylation / cytoplasm protein quality control / aggresome assembly / ubiquitin-modified protein reader activity / regulation of protein localization to chromatin / cellular response to misfolded protein / mitotic spindle disassembly / VCP-NPL4-UFD1 AAA ATPase complex / positive regulation of mitochondrial membrane potential / vesicle-fusing ATPase / K48-linked polyubiquitin modification-dependent protein binding / ciliary transition zone / regulation of aerobic respiration / NAD+ metabolic process / retrograde protein transport, ER to cytosol / stress granule disassembly / ubiquitin-specific protease binding / regulation of synapse organization / positive regulation of ATP biosynthetic process / intracellular membrane-bounded organelle / ubiquitin-like protein ligase binding / RHOH GTPase cycle / MHC class I protein binding / autophagosome maturation / negative regulation of hippo signaling / endoplasmic reticulum to Golgi vesicle-mediated transport / HSF1 activation / polyubiquitin modification-dependent protein binding / interstrand cross-link repair / ATP metabolic process / Attachment and Entry / endoplasmic reticulum unfolded protein response / Protein methylation / ERAD pathway / ciliary tip / translesion synthesis / negative regulation of protein localization to chromatin / lipid droplet / viral genome replication / proteasome complex / macroautophagy / negative regulation of smoothened signaling pathway / Josephin domain DUBs / establishment of protein localization / proteasomal protein catabolic process / N-glycan trimming in the ER and Calnexin/Calreticulin cycle / positive regulation of protein-containing complex assembly / ADP binding / Hh mutants are degraded by ERAD / positive regulation of non-canonical NF-kappaB signal transduction / Translesion Synthesis by POLH / Hedgehog ligand biogenesis / Defective CFTR causes cystic fibrosis / ABC-family protein mediated transport / autophagy / cytoplasmic stress granule / positive regulation of protein catabolic process / positive regulation of canonical Wnt signaling pathway / Aggrephagy / azurophil granule lumen / Ovarian tumor domain proteases / KEAP1-NFE2L2 pathway / double-strand break repair / positive regulation of proteasomal ubiquitin-dependent protein catabolic process / cellular response to heat / E3 ubiquitin ligases ubiquitinate target proteins / site of double-strand break / Neddylation / secretory granule lumen / protein phosphatase binding / regulation of apoptotic process / ficolin-1-rich granule lumen / ciliary basal body / ubiquitin-dependent protein catabolic process / proteasome-mediated ubiquitin-dependent protein catabolic process / Attachment and Entry / protein ubiquitination / protein domain specific binding / signaling receptor binding / DNA repair / ubiquitin protein ligase binding / Neutrophil degranulation / DNA damage response / lipid binding / endoplasmic reticulum membrane / perinuclear region of cytoplasm / glutamatergic synapse Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 2.4 Å | |||||||||
 Authors Authors | Banerjee S / Bartesaghi A / Merk A / Rao P / Bulfer SL / Yan Y / Green N / Mroczkowski B / Neitz RJ / Wipf P ...Banerjee S / Bartesaghi A / Merk A / Rao P / Bulfer SL / Yan Y / Green N / Mroczkowski B / Neitz RJ / Wipf P / Falconieri V / Deshaies RJ / Milne JLS / Huryn D / Arkin M / Subramaniam S | |||||||||
 Citation Citation | Journal: Science / Year: 2016 Title: 2.3 Å resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Authors: Soojay Banerjee / Alberto Bartesaghi / Alan Merk / Prashant Rao / Stacie L Bulfer / Yongzhao Yan / Neal Green / Barbara Mroczkowski / R Jeffrey Neitz / Peter Wipf / Veronica Falconieri / ...Authors: Soojay Banerjee / Alberto Bartesaghi / Alan Merk / Prashant Rao / Stacie L Bulfer / Yongzhao Yan / Neal Green / Barbara Mroczkowski / R Jeffrey Neitz / Peter Wipf / Veronica Falconieri / Raymond J Deshaies / Jacqueline L S Milne / Donna Huryn / Michelle Arkin / Sriram Subramaniam /  Abstract: p97 is a hexameric AAA+ adenosine triphosphatase (ATPase) that is an attractive target for cancer drug development. We report cryo-electron microscopy (cryo-EM) structures for adenosine diphosphate ...p97 is a hexameric AAA+ adenosine triphosphatase (ATPase) that is an attractive target for cancer drug development. We report cryo-electron microscopy (cryo-EM) structures for adenosine diphosphate (ADP)-bound, full-length, hexameric wild-type p97 in the presence and absence of an allosteric inhibitor at resolutions of 2.3 and 2.4 angstroms, respectively. We also report cryo-EM structures (at resolutions of ~3.3, 3.2, and 3.3 angstroms, respectively) for three distinct, coexisting functional states of p97 with occupancies of zero, one, or two molecules of adenosine 5'-O-(3-thiotriphosphate) (ATPγS) per protomer. A large corkscrew-like change in molecular architecture, coupled with upward displacement of the N-terminal domain, is observed only when ATPγS is bound to both the D1 and D2 domains of the protomer. These cryo-EM structures establish the sequence of nucleotide-driven structural changes in p97 at atomic resolution. They also enable elucidation of the binding mode of an allosteric small-molecule inhibitor to p97 and illustrate how inhibitor binding at the interface between the D1 and D2 domains prevents propagation of the conformational changes necessary for p97 function. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_3296.map.gz | 106.3 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-3296-v30.xmlemd-3296.xml | 11.2 KB 11.2 KB | Display Display | EMDB header |
| Images |  emd_3296.png emd_3296.png | 241.1 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-3296ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3296 http://ftp.pdbj.org/pub/emdb/structures/EMD-3296ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3296 | HTTPS FTP |
-Related structure data
| Related structure data |  5ftkMC  3295C  3297C  3298C  3299C  5ftjC  5ftlC  5ftmC  5ftnC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |














-Map
| File | Download / File: emd_3296.map.gz / Format: CCP4 / Size: 113.1 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Reconstruction of human p97 bound to ADP. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.637 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : Full-length human p97
| Entire | Name: Full-length human p97 |
|---|---|
| Components |
|
-Supramolecule #1000: Full-length human p97
| Supramolecule | Name: Full-length human p97 / type: sample / ID: 1000 / Details: The sample was monodisperse. / Oligomeric state: hexamer / Number unique components: 1 |
|---|---|
| Molecular weight | Theoretical: 540 KDa / Method: sequence |
-Macromolecule #1: p97/VCP Transitional endoplasmic reticulum ATPase
| Macromolecule | Name: p97/VCP Transitional endoplasmic reticulum ATPase / type: protein_or_peptide / ID: 1 / Name.synonym: p97 / Number of copies: 1 / Oligomeric state: hexamer / Recombinant expression: Yes |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) / synonym: Human / Location in cell: cytoplasm |
| Molecular weight | Theoretical: 540 KDa |
| Recombinant expression | Organism: Bacteria (eubacteria) / Recombinant cell: E. coli / Recombinant plasmid: Rosetta2 (DE3) |
| Sequence | UniProtKB: Transitional endoplasmic reticulum ATPase GO: ATP binding, signaling receptor binding, protein binding, lipid binding |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.9 mg/mL |
|---|---|
| Buffer | pH: 8 / Details: 25 mM Tris, 150 mM NaCl, 1 mM MgCl2, 1.0 mM TCEP |
| Grid | Details: 200 mesh Quantifoil R1.2/1/3 grids |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 90.15 K / Instrument: FEI VITROBOT MARK IV / Method: Blot for 2.5 seconds before plunging. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Temperature | Min: 79.6 K / Max: 79.8 K / Average: 79.7 K |
| Specialist optics | Energy filter - Name: Gatan, Inc. / Energy filter - Lower energy threshold: 0.0 eV / Energy filter - Upper energy threshold: 20.0 eV |
| Details | Parallel beam illumination |
| Date | Sep 19, 2015 |
| Image recording | Category: CCD / Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Number real images: 1925 / Average electron dose: 40 e/Å2 Details: Every image is the average of 38 frames recorded by the direct electron detector. |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Calibrated magnification: 78490 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.5 µm / Nominal defocus min: 0.7 µm / Nominal magnification: 215000 |
| Sample stage | Specimen holder: Liquid nitrogen cooled / Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| CTF correction | Details: each particle |
|---|---|
| Final reconstruction | Applied symmetry - Point group: C6 (6 fold cyclic) / Algorithm: OTHER / Resolution.type: BY AUTHOR / Resolution: 2.4 Å / Resolution method: OTHER / Software - Name: FREALIGN / Details: Map was corrected using a B-factor of -80 A^2. / Number images used: 30616 |
