 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-17232: Purified human apoferritin at 2.8 A resolution from tilt series p... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Purified human apoferritin at 2.8 A resolution from tilt series processed with TomoBEAR | |||||||||||||||
 Map data Map data | Final map, filtered according to local resolution in RELION, sharpened with a B factor of -70. Nominal apix is 1.378, but was calibrated to 1.331. | |||||||||||||||
 Sample Sample |
| |||||||||||||||
 Keywords Keywords | apoferritin / heavy chain / purified sample / StA / tomography / cryo-ET / TomoBEAR / tilt series / CAFR / automated processing / benchmark / OXIDOREDUCTASE | |||||||||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||||||||
| Method | subtomogram averaging / cryo EM / Resolution: 2.8 Å | |||||||||||||||
 Authors Authors | Balyschew N / Yushkevich A / Mikirtumov V / Sanchez RM / Sprink T / Kudryashev M | |||||||||||||||
| Funding support |  Germany, 4 items Germany, 4 items
| |||||||||||||||
 Citation Citation | Journal: Nat Commun / Year: 2023 Title: Streamlined structure determination by cryo-electron tomography and subtomogram averaging using TomoBEAR. Authors: Nikita Balyschew / Artsemi Yushkevich / Vasilii Mikirtumov / Ricardo M Sanchez / Thiemo Sprink / Mikhail Kudryashev / Abstract: Structures of macromolecules in their native state provide unique unambiguous insights into their functions. Cryo-electron tomography combined with subtomogram averaging demonstrated the power to ...Structures of macromolecules in their native state provide unique unambiguous insights into their functions. Cryo-electron tomography combined with subtomogram averaging demonstrated the power to solve such structures in situ at resolutions in the range of 3 Angstrom for some macromolecules. In order to be applicable to the structural determination of the majority of macromolecules observable in cells in limited amounts, processing of tomographic data has to be performed in a high-throughput manner. Here we present TomoBEAR-a modular configurable workflow engine for streamlined processing of cryo-electron tomographic data for subtomogram averaging. TomoBEAR combines commonly used cryo-EM packages with reasonable presets to provide a transparent ("white box") approach for data management and processing. We demonstrate applications of TomoBEAR to two data sets of purified macromolecular targets, to an ion channel RyR1 in a membrane, and the tomograms of plasma FIB-milled lamellae and demonstrate the ability to produce high-resolution structures. TomoBEAR speeds up data processing, minimizes human interventions, and will help accelerate the adoption of in situ structural biology by cryo-ET. The source code and the documentation are freely available. #1: Journal: bioRxiv / Year: 2023Title: Streamlined Structure Determination by Cryo-Electron Tomography and Subtomogram Averaging using TomoBEAR Authors: Balyschew N / Yushkevich A / Mikirtumov V / Sanchez RM / Sprink T / Kudryashev M | |||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|
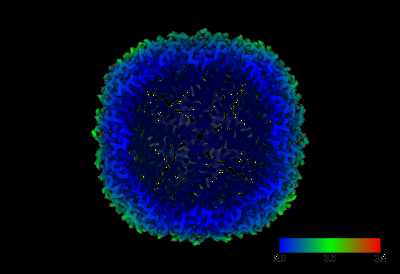
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_17232.map.gz | 19.5 MB |  EMDB map data format EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-17232-v30.xmlemd-17232.xml | 16.3 KB 16.3 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_17232_fsc.xml | 7.1 KB | Display | FSC data file |
| Images | 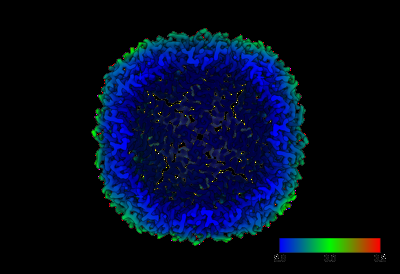 emd_17232.png emd_17232.png | 73.2 KB | ||
| Masks | emd_17232_msk_1.map | 30.5 MB | Mask map | |
| Filedesc metadata | emd-17232.cif.gz | 4.7 KB | ||
| Others | emd_17232_half_map_1.map.gzemd_17232_half_map_2.map.gz | 15.1 MB 15.1 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-17232ftp://ftp.pdbj.org/pub/emdb/structures/EMD-17232 http://ftp.pdbj.org/pub/emdb/structures/EMD-17232ftp://ftp.pdbj.org/pub/emdb/structures/EMD-17232 | HTTPS FTP |
-Related structure data


-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_17232.map.gz / Format: CCP4 / Size: 30.5 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Final map, filtered according to local resolution in RELION, sharpened with a B factor of -70. Nominal apix is 1.378, but was calibrated to 1.331. | ||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.331 Å | ||||||||||||||||||||||||||||||||||||
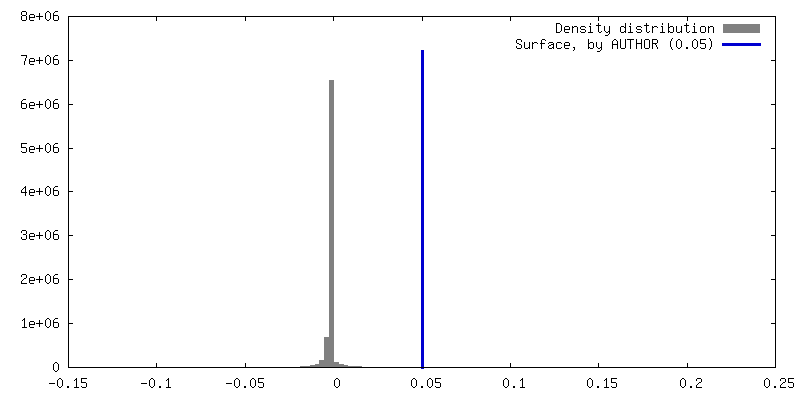
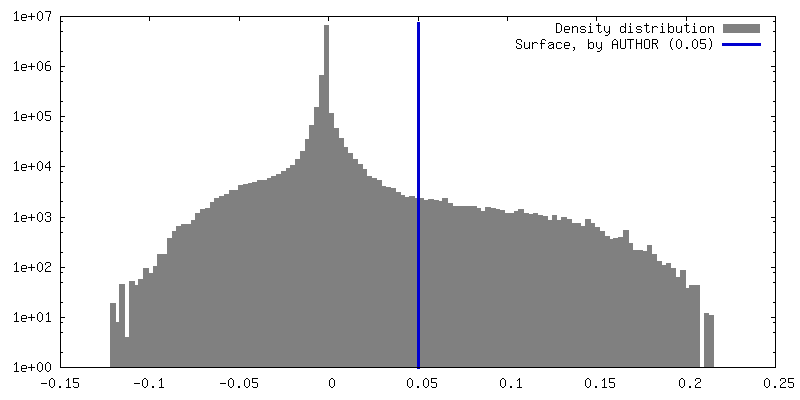
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)



















-Supplemental data
-Mask #1
| File | emd_17232_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
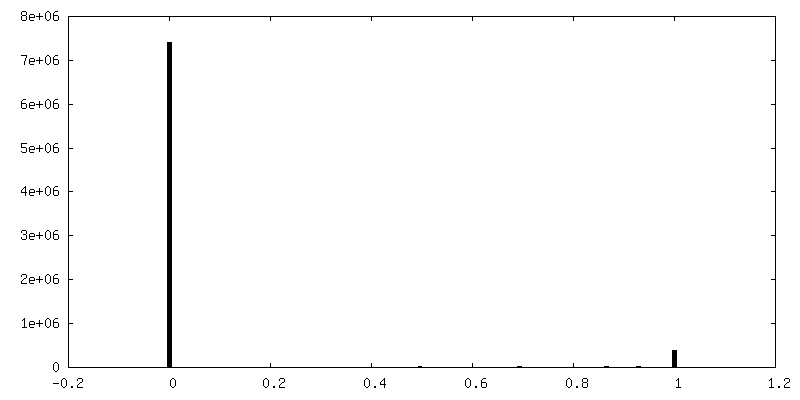
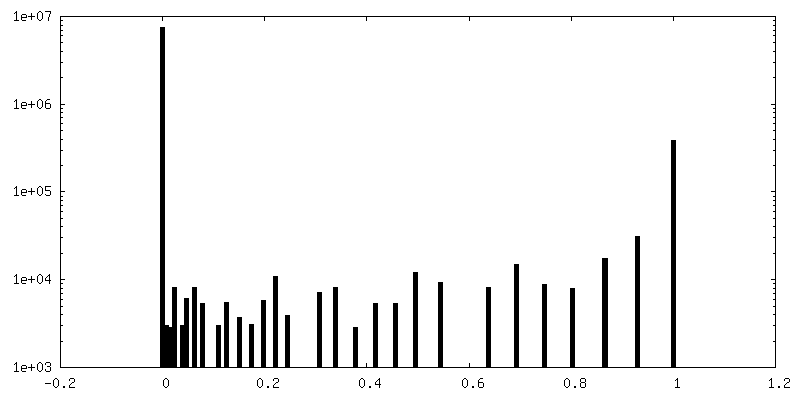
| Density Histograms |






-Half map: #2
| File | emd_17232_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
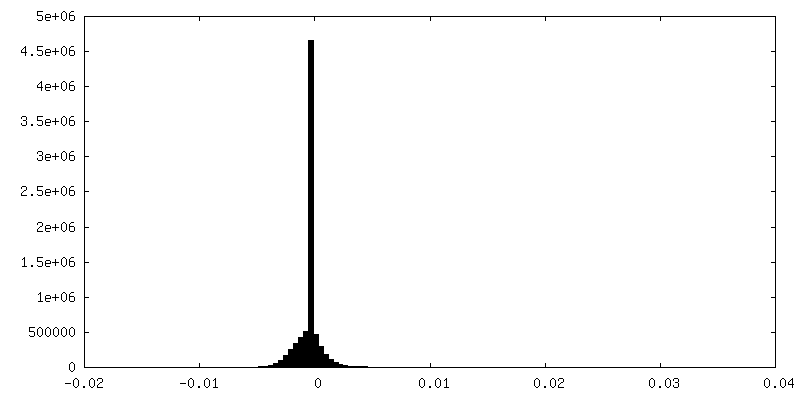
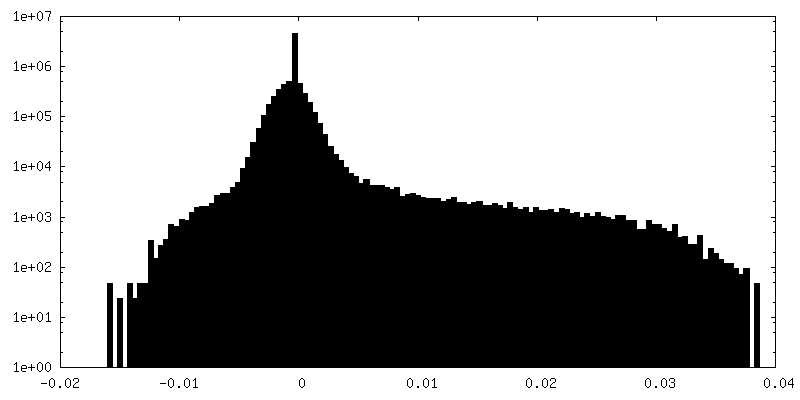
| Density Histograms |






-Half map: #1
| File | emd_17232_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |






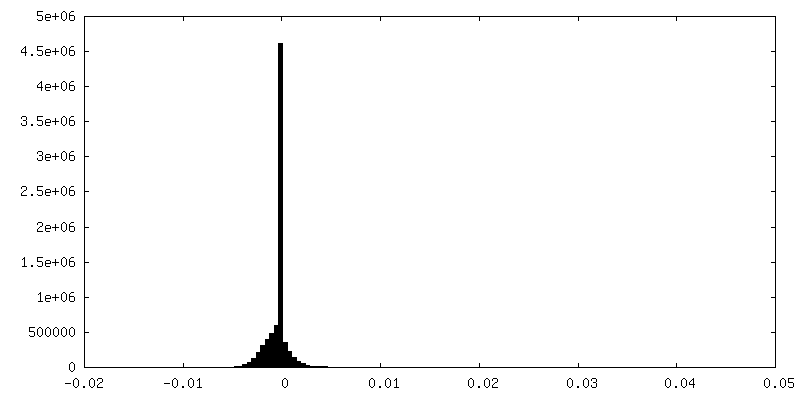
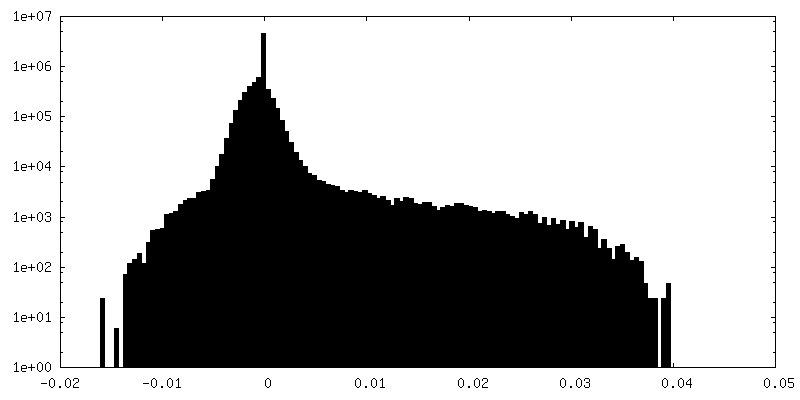
- Sample components
Sample components
-Entire : Apoferritin
| Entire | Name: Apoferritin |
|---|---|
| Components |
|
-Supramolecule #1: Apoferritin
| Supramolecule | Name: Apoferritin / type: organelle_or_cellular_component / ID: 1 / Parent: 0 |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | subtomogram averaging |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 7 |
|---|---|
| Grid | Model: Quantifoil R1.2/1.3 / Material: GOLD |
| Vitrification | Cryogen name: ETHANE / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Specialist optics | Energy filter - Name: GIF Bioquantum / Energy filter - Slit width: 20 eV |
| Image recording | Film or detector model: GATAN K3 (6k x 4k) / Number real images: 1 / Average electron dose: 3.67 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Calibrated defocus max: 5.0 µm / Calibrated defocus min: 2.0 µm / Calibrated magnification: 64000 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: -5.0 µm / Nominal defocus min: -2.0 µm / Nominal magnification: 64000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Final reconstruction | Number classes used: 1 / Applied symmetry - Point group: O (octahedral) / Algorithm: FOURIER SPACE / Resolution.type: BY AUTHOR / Resolution: 2.8 Å / Resolution method: FSC 0.143 CUT-OFF / Software - Name: RELION (ver. 4.0beta) / Software - details: 4.0-beta-2-commit-ce2e93 / Number subtomograms used: 21068 |
|---|---|
| Extraction | Number tomograms: 60 / Number images used: 21886 Reference model: bagel with dimensions similar to apoferritin Method: Template Matching / Software - Name: Dynamo (ver. 0.2.0) Software - details: Dynamo template matching implemented in TomoBEAR with GPU utilization |
| Final angle assignment | Type: MAXIMUM LIKELIHOOD / Software - Name: RELION (ver. 4.0beta) / Software - details: 4.0-beta-2-commit-ce2e93 |
| FSC plot (resolution estimation) | 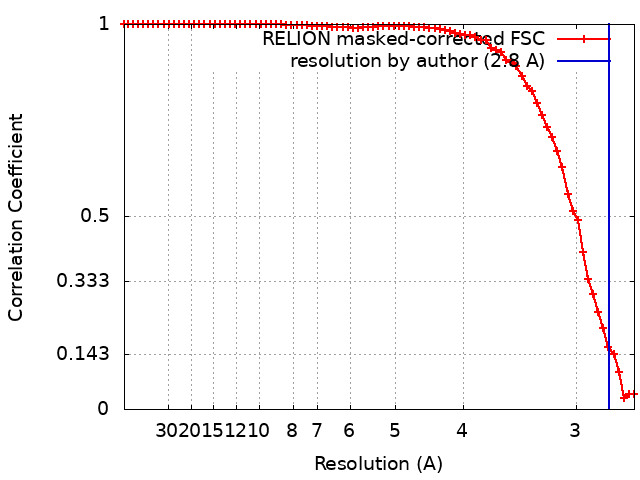 |
