Journal: J Vis Exp / Year: 2022 Title: Routine Collection of High-Resolution cryo-EM Datasets Using 200 KV Transmission Electron Microscope. Authors: Adrian Koh / Sagar Khavnekar / Wen Yang / Dimple Karia / Dennis Cats / Rob van der Ploeg / Fanis Grollios / Oliver Raschdorf / Abhay Kotecha / Daniel Němeček / Abstract: Cryo-electron microscopy (cryo-EM) has been established as a routine method for protein structure determination during the past decade, taking an ever-increasing share of published structural data. ...Cryo-electron microscopy (cryo-EM) has been established as a routine method for protein structure determination during the past decade, taking an ever-increasing share of published structural data. Recent advances in TEM technology and automation have boosted both the speed of data collection and quality of acquired images while simultaneously decreasing the required level of expertise for obtaining cryo-EM maps at sub-3 Å resolutions. While most of such high-resolution structures have been obtained using state-of-the-art 300 kV cryo-TEM systems, high-resolution structures can be also obtained with 200 kV cryo-TEM systems, especially when equipped with an energy filter. Additionally, automation of microscope alignments and data collection with real-time image quality assessment reduces system complexity and assures optimal microscope settings, resulting in increased yield of high-quality images and overall throughput of data collection. This protocol demonstrates the implementation of recent technological advances and automation features on a 200 kV cryo-transmission electron microscope and shows how to collect data for the reconstruction of 3D maps that are sufficient for de novo atomic model building. We focus on best practices, critical variables, and common issues that must be considered to enable the routine collection of such high-resolution cryo-EM datasets. Particularly the following essential topics are reviewed in detail: i) automation of microscope alignments, ii) selection of suitable areas for data acquisition, iii) optimal optical parameters for high-quality, high-throughput data collection, iv) energy filter tuning for zero-loss imaging, and v) data management and quality assessment. Application of the best practices and improvement of achievable resolution using an energy filter will be demonstrated on the example of apo-ferritin that was reconstructed to 1.6 Å, and Thermoplasma acidophilum 20S proteasome reconstructed to 2.1-Å resolution using a 200 kV TEM equipped with an energy filter and a direct electron detector.
History
Deposition
Feb 28, 2022
-
Header (metadata) release
Mar 9, 2022
-
Map release
Mar 9, 2022
-
Update
Dec 13, 2023
-
Current status
Dec 13, 2023
Processing site: PDBe / Status: Released
-
Structure visualization
Movie
Surface view with section colored by density value
EMPIAR-10976 (Title: 2.1Å T20S Proteosome from 200kV Glacios with Selectris Falcon 4 Data size: 1.5 TB Data #1: Unaligned multiframe EER movies of T20S from Falcon4 [micrographs - multiframe])
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Map data
Map data Sample
Sample Keywords
Keywords Function and homology information
Function and homology information
 Thermoplasma acidophilum (acidophilic)
Thermoplasma acidophilum (acidophilic) Authors
Authors Netherlands, 1 items
Netherlands, 1 items  Citation
Citation
 Structure visualization
Structure visualization
 Downloads & links
Downloads & links emd_14467.png
emd_14467.png http://ftp.pdbj.org/pub/emdb/structures/EMD-14467
http://ftp.pdbj.org/pub/emdb/structures/EMD-14467












 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)
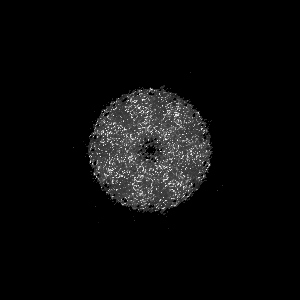







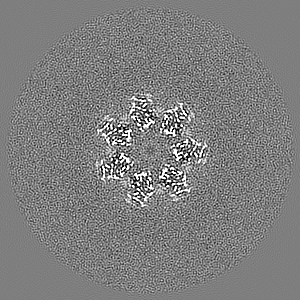




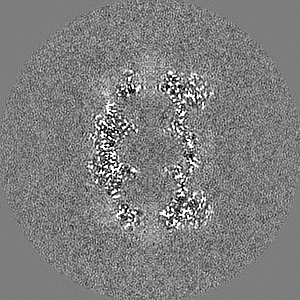




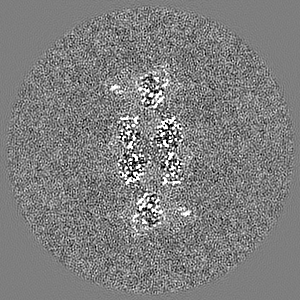








 Sample components
Sample components Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN
