 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of Rep protein in SaPI1 | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
| Biological species |   Staphylococcus aureus (bacteria) Staphylococcus aureus (bacteria) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.9 Å | |||||||||
 Authors Authors | Qiao CC / Mir-Sanchis I | |||||||||
| Funding support |  Sweden, 1 items Sweden, 1 items
| |||||||||
 Citation Citation | Journal: Nucleic Acids Res / Year: 2022 Title: Staphylococcal self-loading helicases couple the staircase mechanism with inter domain high flexibility. Authors: Cuncun Qiao / Gianluca Debiasi-Anders / Ignacio Mir-Sanchis / Abstract: Replication is a crucial cellular process. Replicative helicases unwind DNA providing the template strand to the polymerase and promoting replication fork progression. Helicases are multi-domain ...Replication is a crucial cellular process. Replicative helicases unwind DNA providing the template strand to the polymerase and promoting replication fork progression. Helicases are multi-domain proteins which use an ATPase domain to couple ATP hydrolysis with translocation, however the role that the other domains might have during translocation remains elusive. Here, we studied the unexplored self-loading helicases called Reps, present in Staphylococcus aureus pathogenicity islands (SaPIs). Our cryoEM structures of the PriRep5 from SaPI5 (3.3 Å), the Rep1 from SaPI1 (3.9 Å) and Rep1-DNA complex (3.1Å) showed that in both Reps, the C-terminal domain (CTD) undergoes two distinct movements respect the ATPase domain. We experimentally demonstrate both in vitro and in vivo that SaPI-encoded Reps need key amino acids involved in the staircase mechanism of translocation. Additionally, we demonstrate that the CTD's presence is necessary for the maintenance of full ATPase and helicase activities. We speculate that this high interdomain flexibility couples Rep's activities as initiators and as helicases. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_12987.map.gz | 58.8 MB |  EMDB map data format EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-12987-v30.xmlemd-12987.xml | 14.5 KB 14.5 KB | Display Display | EMDB header |
| Images | 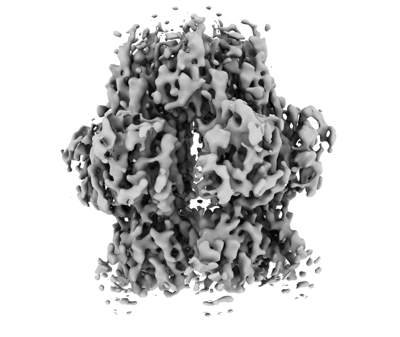 emd_12987.png emd_12987.png | 86 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-12987ftp://ftp.pdbj.org/pub/emdb/structures/EMD-12987 http://ftp.pdbj.org/pub/emdb/structures/EMD-12987ftp://ftp.pdbj.org/pub/emdb/structures/EMD-12987 | HTTPS FTP |
-Related structure data
| Related structure data |  7olaC  7om0C  7pdsC C: citing same article ( |
|---|---|
| EM raw data | EMPIAR-10945 (Title: Staphylococcal self-loading helicases couple the staircase mechanism 1 with inter domain high flexibility Data size: 1.6 TB / Data #1: Rep1apo [micrographs - multiframe]) |




-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_12987.map.gz / Format: CCP4 / Size: 64 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.82 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




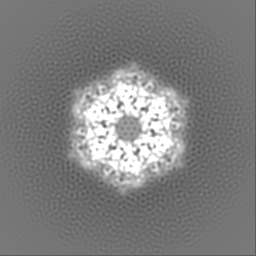







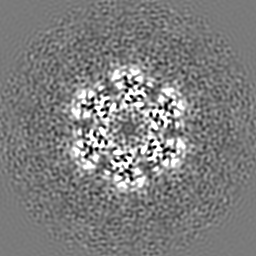








-Supplemental data
- Sample components
Sample components
-Entire : Rep1 in SaPI1
| Entire | Name: Rep1 in SaPI1 |
|---|---|
| Components |
|
-Supramolecule #1: Rep1 in SaPI1
| Supramolecule | Name: Rep1 in SaPI1 / type: organelle_or_cellular_component / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Staphylococcus aureus (bacteria) / Strain: U93688 |
| Molecular weight | Experimental: 330 KDa |
| Recombinant expression | Organism: |
-Macromolecule #1: Rep1
| Macromolecule | Name: Rep1 / type: protein_or_peptide / ID: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Staphylococcus aureus (bacteria) / Strain: U93688 |
| Recombinant expression | Organism: |
| Sequence | String: GSHMFEMIDS RTGVLNANDW KSQLRRSATT QALKKTTTNA EIILCNDESL KGLVQYDAFE KVTKLKRLPY WRSKGDANYY WADIDTTHVI SHIDKLYNVQ FSRDLIDTVI EKEAYQNRFH PIKSMIESKS WDGIKRIETL FIDYLGAEDN HYNREVTKKW MMGAVARIYQ ...String: GSHMFEMIDS RTGVLNANDW KSQLRRSATT QALKKTTTNA EIILCNDESL KGLVQYDAFE KVTKLKRLPY WRSKGDANYY WADIDTTHVI SHIDKLYNVQ FSRDLIDTVI EKEAYQNRFH PIKSMIESKS WDGIKRIETL FIDYLGAEDN HYNREVTKKW MMGAVARIYQ PGIKYDSMII LYGGQGVGKS TAVSKLGGHW YNQSIKTFKG DEVYKKLQGS WICEIEELSA FQKSTIEDIK GFISAIVDIY RASYGKRTER HPRQCVFVGT TNNYEFLKDQ TGNRRFFPIT TDKNKATKSP FDDLTPVVVQ QMFAEARVYF DENPTDKALL LDKEASEMAL KVQEAHSEKD ALVGEIEEFL ERPIPSDYWY RTLEEKRVSA HDVIDQDYIK LYGDGKLIEL PNAKPGAYVW RDKVCSMEIW KVMMKRDDQP QQHHLRKIDK ALRNTNYCGT VKKQTRYGEG IGKQYGFSVD LASYYKNLKV |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.2 mg/mL | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 8 Component:
Details: 20mMTris pH 8, 200 mM NaCl, 0.5mM EDTA, 2mM DTT | |||||||||||||||
| Grid | Model: Quantifoil R2/2 / Material: COPPER / Mesh: 200 / Support film - Material: CARBON / Support film - topology: CONTINUOUS / Support film - Film thickness: 2.0 nm / Pretreatment - Type: GLOW DISCHARGE | |||||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy #1
Electron microscopy #1
| Microscopy ID | 1 |
|---|---|
| Microscope | FEI TITAN KRIOS |
| Image recording | Image recording ID: 1 / Film or detector model: GATAN K2 QUANTUM (4k x 4k) / Detector mode: COUNTING / Number real images: 3345 / Average exposure time: 4.0 sec. / Average electron dose: 1.08 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 100.0 µm / Illumination mode: SPOT SCAN / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.4 µm / Nominal defocus min: 1.2 µm / Nominal magnification: 165000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Electron microscopy #1~
| Microscopy ID | 1 |
|---|---|
| Microscope | FEI TITAN KRIOS |
| Image recording | Image recording ID: 2 / Film or detector model: GATAN K2 QUANTUM (4k x 4k) / Detector mode: COUNTING / Number real images: 1158 / Average exposure time: 4.0 sec. / Average electron dose: 1.08 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 100.0 µm / Illumination mode: SPOT SCAN / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.4 µm / Nominal defocus min: 1.2 µm / Nominal magnification: 165000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment | Model: Titan Krios / Image courtesy: FEI Company |
