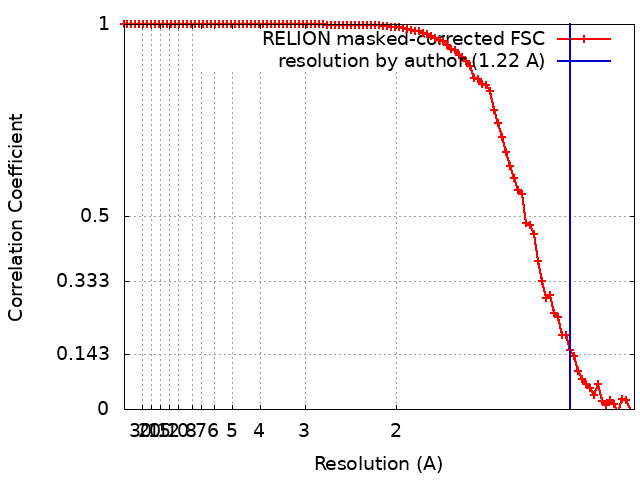
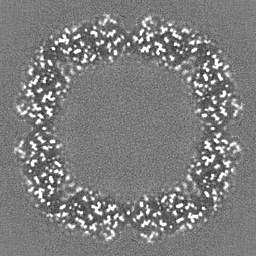
Journal: Nature / Year: 2020 Title: Single-particle cryo-EM at atomic resolution. Authors: Takanori Nakane / Abhay Kotecha / Andrija Sente / Greg McMullan / Simonas Masiulis / Patricia M G E Brown / Ioana T Grigoras / Lina Malinauskaite / Tomas Malinauskas / Jonas Miehling / ...Authors: Takanori Nakane / Abhay Kotecha / Andrija Sente / Greg McMullan / Simonas Masiulis / Patricia M G E Brown / Ioana T Grigoras / Lina Malinauskaite / Tomas Malinauskas / Jonas Miehling / Tomasz Uchański / Lingbo Yu / Dimple Karia / Evgeniya V Pechnikova / Erwin de Jong / Jeroen Keizer / Maarten Bischoff / Jamie McCormack / Peter Tiemeijer / Steven W Hardwick / Dimitri Y Chirgadze / Garib Murshudov / A Radu Aricescu / Sjors H W Scheres / Abstract: The three-dimensional positions of atoms in protein molecules define their structure and their roles in biological processes. The more precisely atomic coordinates are determined, the more chemical ...The three-dimensional positions of atoms in protein molecules define their structure and their roles in biological processes. The more precisely atomic coordinates are determined, the more chemical information can be derived and the more mechanistic insights into protein function may be inferred. Electron cryo-microscopy (cryo-EM) single-particle analysis has yielded protein structures with increasing levels of detail in recent years. However, it has proved difficult to obtain cryo-EM reconstructions with sufficient resolution to visualize individual atoms in proteins. Here we use a new electron source, energy filter and camera to obtain a 1.7 Å resolution cryo-EM reconstruction for a human membrane protein, the β3 GABA receptor homopentamer. Such maps allow a detailed understanding of small-molecule coordination, visualization of solvent molecules and alternative conformations for multiple amino acids, and unambiguous building of ordered acidic side chains and glycans. Applied to mouse apoferritin, our strategy led to a 1.22 Å resolution reconstruction that offers a genuine atomic-resolution view of a protein molecule using single-particle cryo-EM. Moreover, the scattering potential from many hydrogen atoms can be visualized in difference maps, allowing a direct analysis of hydrogen-bonding networks. Our technological advances, combined with further approaches to accelerate data acquisition and improve sample quality, provide a route towards routine application of cryo-EM in high-throughput screening of small molecule modulators and structure-based drug discovery.
History
Deposition
Aug 20, 2020
-
Header (metadata) release
Oct 28, 2020
-
Map release
Oct 28, 2020
-
Update
Jul 10, 2024
-
Current status
Jul 10, 2024
Processing site: PDBe / Status: Released
-
Structure visualization
Movie
Surface view with section colored by density value
EMPIAR-10424 (Title: Atomic resolution structure of apoferritin / Data size: 538.1 Data #1: Unaligned movies of apoferritin [micrographs - multiframe])
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Map data
Map data Sample
Sample Keywords
Keywords Function and homology information
Function and homology information
 Authors
Authors United Kingdom, 6 items
United Kingdom, 6 items  Citation
Citation

 Structure visualization
Structure visualization
 Downloads & links
Downloads & links emd_11638.png
emd_11638.png http://ftp.pdbj.org/pub/emdb/structures/EMD-11638
http://ftp.pdbj.org/pub/emdb/structures/EMD-11638



























 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)






























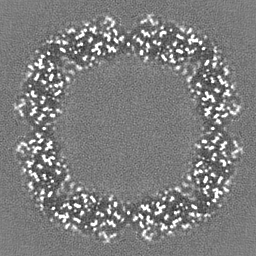
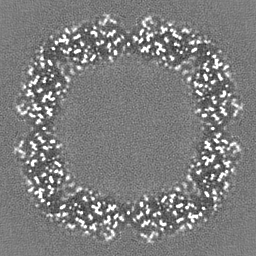



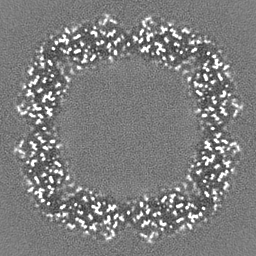
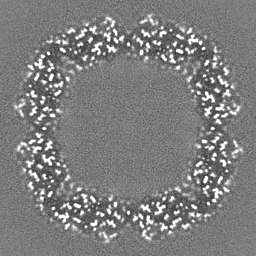
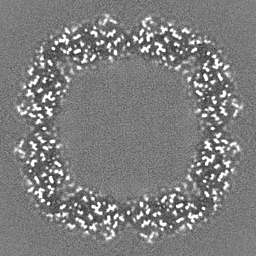
 Sample components
Sample components

 Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN