 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1aj6: NOVOBIOCIN-RESISTANT MUTANT (R136H) OF THE N-TERMINAL 24 KDA FRAG... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1aj6 | ||||||
|---|---|---|---|---|---|---|---|
| Title | NOVOBIOCIN-RESISTANT MUTANT (R136H) OF THE N-TERMINAL 24 KDA FRAGMENT OF DNA GYRASE B COMPLEXED WITH NOVOBIOCIN AT 2.3 ANGSTROMS RESOLUTION | ||||||
 Components Components | GYRASE | ||||||
 Keywords Keywords | TOPOISOMERASE / GYRASE / NOVOBIOCIN / ANTIBIOTIC / RESISTANT MUTANT | ||||||
| Function / homology |  Function and homology information Function and homology informationAction of antimicrobials / DNA topoisomerase type II (double strand cut, ATP-hydrolyzing) complex / DNA negative supercoiling activity / DNA topoisomerase type II (double strand cut, ATP-hydrolyzing) activity / DNA topoisomerase (ATP-hydrolysing) / Antimicrobial resistance / DNA topological change / ATP-dependent activity, acting on DNA / DNA-templated DNA replication / chromosome ...Action of antimicrobials / DNA topoisomerase type II (double strand cut, ATP-hydrolyzing) complex / DNA negative supercoiling activity / DNA topoisomerase type II (double strand cut, ATP-hydrolyzing) activity / DNA topoisomerase (ATP-hydrolysing) / Antimicrobial resistance / DNA topological change / ATP-dependent activity, acting on DNA / DNA-templated DNA replication / chromosome / response to xenobiotic stimulus / response to antibiotic / DNA-templated transcription / DNA binding / ATP binding / metal ion binding / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / DIFFERENCE FOURIER / Resolution: 2.3 Å X-RAY DIFFRACTION / SYNCHROTRON / DIFFERENCE FOURIER / Resolution: 2.3 Å | ||||||
 Authors Authors | Weston, S.A. / Tunnicliffe, A. / Pauptit, R.A. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1997 Title: The entropic penalty of ordered water accounts for weaker binding of the antibiotic novobiocin to a resistant mutant of DNA gyrase: a thermodynamic and crystallographic study. Authors: Holdgate, G.A. / Tunnicliffe, A. / Ward, W.H. / Weston, S.A. / Rosenbrock, G. / Barth, P.T. / Taylor, I.W. / Pauptit, R.A. / Timms, D. #1: Journal: To be PublishedTitle: Antibacterial Design Based on the Structures of Gyrase-Inhibitor Complexes Authors: Pauptit, R.A. / Weston, S.A. / Breeze, A.L. / Derbyshire, D.J. / Tucker, A.D. / Hales, N. / Hollinshead, D. / Timms, D. #2: Journal: Proteins / Year: 1997Title: The High-Resolution Crystal Structure of a 24-kDa Gyrase B Fragment from E. Coli Complexed with One of the Most Potent Coumarin Inhibitors, Clorobiocin Authors: Tsai, F.T. / Singh, O.M. / Skarzynski, T. / Wonacott, A.J. / Weston, S. / Tucker, A. / Pauptit, R.A. / Breeze, A.L. / Poyser, J.P. / O'Brien, R. / Ladbury, J.E. / Wigley, D.B. #3: Journal: Embo J. / Year: 1996Title: The Nature of Inhibition of DNA Gyrase by the Coumarins and the Cyclothialidines Revealed by X-Ray Crystallography Authors: Lewis, R.J. / Singh, O.M. / Smith, C.V. / Skarzynski, T. / Maxwell, A. / Wonacott, A.J. / Wigley, D.B. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1aj6.cif.gz | 54.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1aj6.ent.gz | 38.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1aj6.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/aj/1aj6ftp://data.pdbj.org/pub/pdb/validation_reports/aj/1aj6 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|









-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 24040.936 Da / Num. of mol.: 1 / Fragment: N-TERMINAL 24 KDA / Mutation: R136H Source method: isolated from a genetically manipulated source Source: (gene. exp.) Description: PLASMID PTB382 ENCODING MUTANT DERIVED FROM PAM24 WHICH ENCODES 24 KDA SUBDOMAIN (SUPPLIED BY A. MAXWELL, LEICESTER), WHICH IN TURN WAS DERIVED FROM PAG111 PLASMID WHICH ENCODES FOR GYRB Cellular location: CYTOPLASM / Gene: GYRB / Cellular location (production host): CYTOPLASM / Production host: References: UniProt: P06982, UniProt: P0AES6*PLUS, EC: 5.99.1.3 |
|---|---|
| #2: Chemical | ChemComp-NOV / 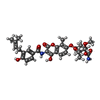  Mass: 612.624 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C31H36N2O11 / Comment: antibiotic*YM Mass: 612.624 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C31H36N2O11 / Comment: antibiotic*YM |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 89 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 89 / Source method: isolated from a natural source / Formula: H2O |
| Compound details | THE MUTATED RESIDUE HIS 136 HAS AN UNUSUAL ECLIPSED SIDE CHAIN CONFORMATION WHICH IS STABILISED BY ...THE MUTATED RESIDUE HIS 136 HAS AN UNUSUAL ECLIPSED SIDE CHAIN CONFORMATI |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.2 Å3/Da / Density % sol: 45 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion, hanging drop / pH: 6.2 Details: HANGING DROP VAPOR DIFFUSION; 12MGS/ML PROTEIN/NOVOBIOCIN MIXED WITH EQUAL VOLUME OF RESERVOIR SOLUTION (12% V/V PEG200, 100MM MES PH6.2), vapor diffusion - hanging drop | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 20 ℃ / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SRS  / Beamline: PX9.6 / Wavelength: 0.87 / Beamline: PX9.6 / Wavelength: 0.87 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jun 1, 1996 / Details: VERTICALLY FOCUSSED PT-COATED MIRROR |
| Radiation | Monochromator: SI(111) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.87 Å / Relative weight: 1 |
| Reflection | Resolution: 2.3→19.8 Å / Num. obs: 9455 / % possible obs: 94.8 % / Observed criterion σ(I): 0 / Redundancy: 5.1 % / Biso Wilson estimate: 23.7 Å2 / Rmerge(I) obs: 0.065 / Rsym value: 0.065 / Net I/σ(I): 8.5 |
| Reflection shell | Resolution: 2.3→2.38 Å / Redundancy: 5.2 % / Rmerge(I) obs: 0.131 / Mean I/σ(I) obs: 5.6 / Rsym value: 0.131 / % possible all: 97 |
| Reflection | *PLUS Num. measured all: 48291 |
| Reflection shell | *PLUS % possible obs: 97 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: DIFFERENCE FOURIER Starting model: WILD-TYPE 24KDA FRAGMENT/NOVOBIOCIN COMPLEX Resolution: 2.3→19.8 Å / Isotropic thermal model: TNT BCORREL / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: TNT PROGEO Details: STRAINED GEOMETRY AND DIFFERENCE DENSITY AT PRO 23 SUGGEST IT MIGHT HAVE MORE THAN ONE CONFORMATION. ASN 178 IS IN A DISALLOWED REGION OF THE RAMACHANDRAN PLOT. IT ADOPTS A CLASSICAL C-7 ...Details: STRAINED GEOMETRY AND DIFFERENCE DENSITY AT PRO 23 SUGGEST IT MIGHT HAVE MORE THAN ONE CONFORMATION. ASN 178 IS IN A DISALLOWED REGION OF THE RAMACHANDRAN PLOT. IT ADOPTS A CLASSICAL C-7 AXIAL CONFORMATION TYPICAL OF GLYCINE RESIDUES WITH A CO(N-1)..N(N+1) H-BOND.
| ||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: TNT / Bsol: 241 Å2 / ksol: 1 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→19.8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: TNT / Version: 5D / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.195 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: t_plane_restr / Dev ideal: 0.015 / Weight: 10 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 2.3 Å / Lowest resolution: 2.4 Å / Rfactor Rfree: 0.132 / Rfactor obs: 0.23 |
