 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-8750 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
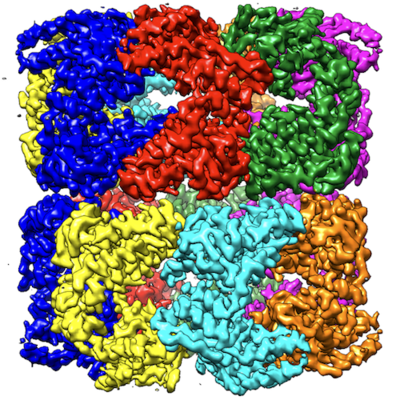
| Title | GroEL using cryoEM | |||||||||
 Map data Map data | GroEL | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | GroEL / cryoEM / conformational heterogeneity. / CHAPERONE | |||||||||
| Function / homology |  Function and homology information Function and homology informationGroEL-GroES complex / chaperonin ATPase / virion assembly / isomerase activity / ATP-dependent protein folding chaperone / response to radiation / : / response to heat / protein refolding / protein folding ...GroEL-GroES complex / chaperonin ATPase / virion assembly / isomerase activity / ATP-dependent protein folding chaperone / response to radiation / : / response to heat / protein refolding / protein folding / magnesium ion binding / ATP hydrolysis activity / ATP binding / membrane / identical protein binding / cytosol / cytoplasm Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.5 Å | |||||||||
 Authors Authors | Roh SH / Chiu W | |||||||||
| Funding support |  United States, 1 items United States, 1 items
| |||||||||
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2017 Title: Subunit conformational variation within individual GroEL oligomers resolved by Cryo-EM. Authors: Soung-Hun Roh / Corey F Hryc / Hyun-Hwan Jeong / Xue Fei / Joanita Jakana / George H Lorimer / Wah Chiu / Abstract: Single-particle electron cryo-microscopy (cryo-EM) is an emerging tool for resolving structures of conformationally heterogeneous particles; however, each structure is derived from an average of many ...Single-particle electron cryo-microscopy (cryo-EM) is an emerging tool for resolving structures of conformationally heterogeneous particles; however, each structure is derived from an average of many particles with presumed identical conformations. We used a 3.5-Å cryo-EM reconstruction with imposed D7 symmetry to further analyze structural heterogeneity among chemically identical subunits in each GroEL oligomer. Focused classification of the 14 subunits in each oligomer revealed three dominant classes of subunit conformations. Each class resembled a distinct GroEL crystal structure in the Protein Data Bank. The conformational differences stem from the orientations of the apical domain. We mapped each conformation class to its subunit locations within each GroEL oligomer in our dataset. The spatial distributions of each conformation class differed among oligomers, and most oligomers contained 10-12 subunits of the three dominant conformation classes. Adjacent subunits were found to more likely assume the same conformation class, suggesting correlation among subunits in the oligomer. This study demonstrates the utility of cryo-EM in revealing structure dynamics within a single protein oligomer. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_8750.map.gz | 49.4 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-8750-v30.xmlemd-8750.xml | 17.1 KB 17.1 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_8750_fsc.xml | 8.5 KB | Display | FSC data file |
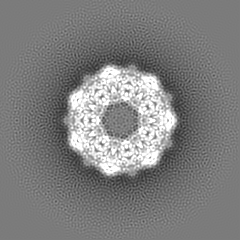
| Images |  emd_8750.png emd_8750.png | 331.9 KB | ||
| Filedesc metadata | emd-8750.cif.gz | 5.7 KB | ||
| Others | emd_8750_additional_1.map.gzemd_8750_additional_2.map.gzemd_8750_additional_3.map.gz | 49.4 MB 49.3 MB 49.4 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-8750ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8750 http://ftp.pdbj.org/pub/emdb/structures/EMD-8750ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8750 | HTTPS FTP |
-Related structure data
| Related structure data |  5w0sMC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |









-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_8750.map.gz / Format: CCP4 / Size: 52.7 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
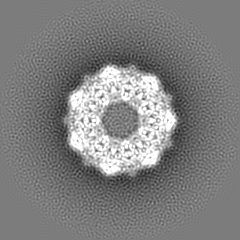
| Annotation | GroEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.23 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
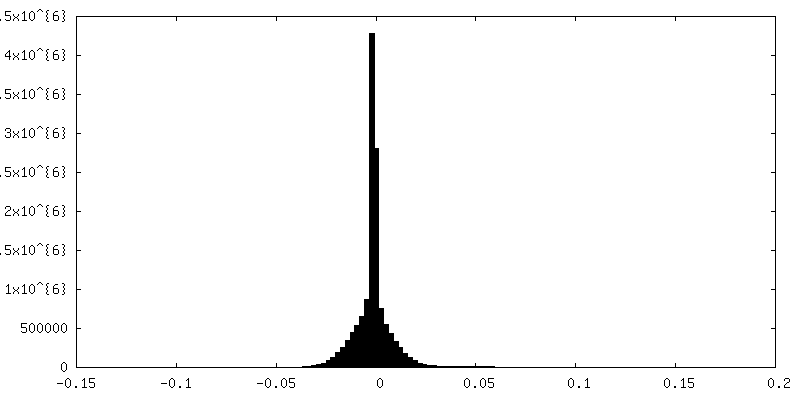
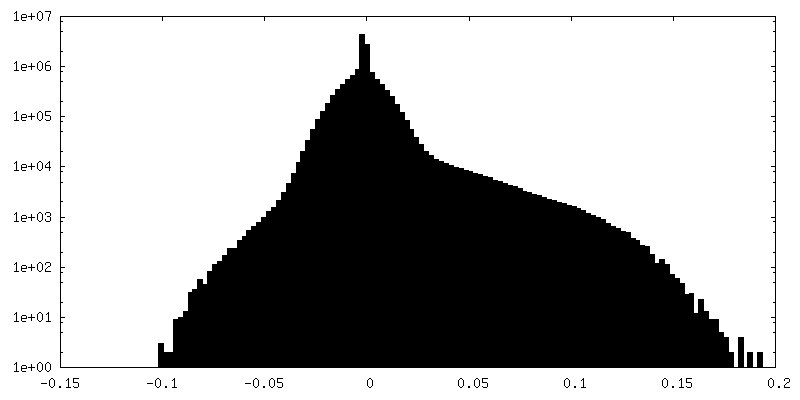
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
-Additional map: GroEL, Conformation 1
| File | emd_8750_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | GroEL, Conformation 1 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |







-Additional map: GroEL, Conformation 2
| File | emd_8750_additional_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | GroEL, Conformation 2 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Additional map: GroEL, Conformation 3
| File | emd_8750_additional_3.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | GroEL, Conformation 3 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |




- Sample components
Sample components
-Entire : Wild type GroEL
| Entire | Name: Wild type GroEL |
|---|---|
| Components |
|
-Supramolecule #1: Wild type GroEL
| Supramolecule | Name: Wild type GroEL / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 800 kDa/nm |
-Macromolecule #1: 60 kDa chaperonin
| Macromolecule | Name: 60 kDa chaperonin / type: protein_or_peptide / ID: 1 / Number of copies: 14 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 55.14802 KDa |
| Recombinant expression | Organism: |
| Sequence | String: AAKDVKFGND AGVKMLRGVN VLADAVKVTL GPKGRNVVLD KSFGAPTITK DGVSVAREIE LEDKFENMGA QMVKEVASKA NDAAGDGTT TATVLAQAII TEGLKAVAAG MNPMDLKRGI DKAVTVAVEE LKALSVPCSD SKAIAQVGTI SANSDETVGK L IAEAMDKV ...String: AAKDVKFGND AGVKMLRGVN VLADAVKVTL GPKGRNVVLD KSFGAPTITK DGVSVAREIE LEDKFENMGA QMVKEVASKA NDAAGDGTT TATVLAQAII TEGLKAVAAG MNPMDLKRGI DKAVTVAVEE LKALSVPCSD SKAIAQVGTI SANSDETVGK L IAEAMDKV GKEGVITVED GTGLQDELDV VEGMQFDRGY LSPYFINKPE TGAVELESPF ILLADKKISN IREMLPVLEA VA KAGKPLL IIAEDVEGEA LATLVVNTMR GIVKVAAVKA PGFGDRRKAM LQDIATLTGG TVISEEIGME LEKATLEDLG QAK RVVINK DTTTIIDGVG EEAAIQGRVA QIRQQIEEAT SDYDREKLQE RVAKLAGGVA VIKVGAATEV EMKEKKARVE DALH ATRAA VEEGVVAGGG VALIRVASKL ADLRGQNEDQ NVGIKVALRA MEAPLRQIVL NCGEEPSVVA NTVKGGDGNY GYNAA TEEY GNMIDMGILD PTKVTRSALQ YAASVAGLMI TTECMVTDLP UniProtKB: Chaperonin GroEL |
-Macromolecule #2: water
| Macromolecule | Name: water / type: ligand / ID: 2 / Number of copies: 107 / Formula: HOH |
|---|---|
| Molecular weight | Theoretical: 18.015 Da |
| Chemical component information |  ChemComp-HOH: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.1 mg/mL |
|---|---|
| Buffer | pH: 7.2 |
| Vitrification | Cryogen name: ETHANE |
- Electron microscopy
Electron microscopy
| Microscope | JEOL 3200FSC |
|---|---|
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Average electron dose: 1.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD |
+Image processing

-Atomic model buiding 1
| Details | Regarding negative occupancies: To assess fit-to-density, we derived cross-correlations at the amino acid level and by means of a map/model FSC. To perform this assessment, we generated a weighted map, derived solely from an atomic model that accounted for both ADP of all atoms and weak/negative density of all charged oxygen atoms, and compared it with the experimental map. The weighted map provides a better approximation of the experimental map by simulating map variability as opposed to treating all atoms equally. The correlations for both the FSC and the per-residue assessment showed improvements when properly weighted, further demonstrating that our model provides a good approximation of the experimental data |
|---|---|
| Output model | PDB-5w0s: |
