Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-8595: CryoEM Structure Refinement by Integrating NMR Chemical Shifts wi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-8595 | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | CryoEM Structure Refinement by Integrating NMR Chemical Shifts with Molecular Dynamics Simulations | |||||||||||||||||||||
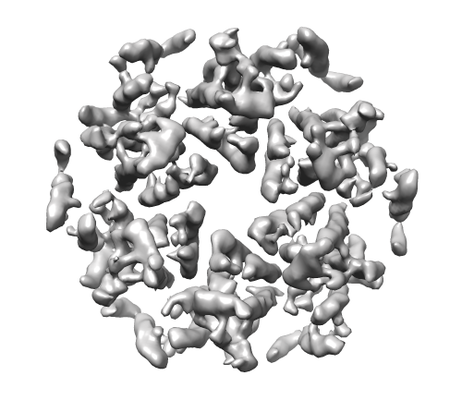
 Map data Map data | HIV-1 CA hexamer | |||||||||||||||||||||
 Sample Sample |
| |||||||||||||||||||||
 Keywords Keywords | Cryo-EM / HIV capsid / Chemical shift / Molecular Dynamics / hydrolase / viral protein | |||||||||||||||||||||
| Function / homology |  Function and homology information Function and homology informationviral budding via host ESCRT complex / host multivesicular body / ISG15 antiviral mechanism / viral nucleocapsid / viral translational frameshifting / host cell nucleus / host cell plasma membrane / virion membrane / structural molecule activity / RNA binding / zinc ion binding Similarity search - Function | |||||||||||||||||||||
| Biological species |  Human immunodeficiency virus type 1 (NEW YORK-5 ISOLATE) Human immunodeficiency virus type 1 (NEW YORK-5 ISOLATE) | |||||||||||||||||||||
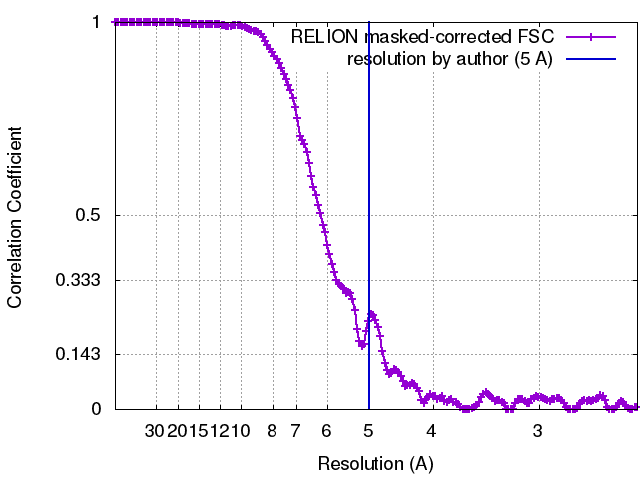
| Method | helical reconstruction / cryo EM / Resolution: 5.0 Å | |||||||||||||||||||||
 Authors Authors | Zhao G / Zhang P | |||||||||||||||||||||
| Funding support |  United States, 6 items United States, 6 items
| |||||||||||||||||||||
 Citation Citation | Journal: J Phys Chem B / Year: 2017 Title: CryoEM Structure Refinement by Integrating NMR Chemical Shifts with Molecular Dynamics Simulations. Authors: Juan R Perilla / Gongpu Zhao / Manman Lu / Jiying Ning / Guangjin Hou / In-Ja L Byeon / Angela M Gronenborn / Tatyana Polenova / Peijun Zhang /  Abstract: Single particle cryoEM has emerged as a powerful method for structure determination of proteins and complexes, complementing X-ray crystallography and NMR spectroscopy. Yet, for many systems, the ...Single particle cryoEM has emerged as a powerful method for structure determination of proteins and complexes, complementing X-ray crystallography and NMR spectroscopy. Yet, for many systems, the resolution of cryoEM density map has been limited to 4-6 Å, which only allows for resolving bulky amino acids side chains, thus hindering accurate model building from the density map. On the other hand, experimental chemical shifts (CS) from solution and solid state MAS NMR spectra provide atomic level data for each amino acid within a molecule or a complex; however, structure determination of large complexes and assemblies based on NMR data alone remains challenging. Here, we present a novel integrated strategy to combine the highly complementary experimental data from cryoEM and NMR computationally by molecular dynamics simulations to derive an atomistic model, which is not attainable by either approach alone. We use the HIV-1 capsid protein (CA) C-terminal domain as well as the large capsid assembly to demonstrate the feasibility of this approach, termed NMR CS-biased cryoEM structure refinement. | |||||||||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_8595.map.gz | 338.4 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-8595-v30.xmlemd-8595.xml | 15 KB 15 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_8595_fsc.xml | 16 KB | Display | FSC data file |
| Images |  emd_8595.png emd_8595.png | 141.6 KB | ||
| Filedesc metadata | emd-8595.cif.gz | 6.1 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-8595ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8595 http://ftp.pdbj.org/pub/emdb/structures/EMD-8595ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8595 | HTTPS FTP |
-Related structure data
| Related structure data |  5upwMC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |








-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |


-Map
| File | Download / File: emd_8595.map.gz / Format: CCP4 / Size: 361.7 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | HIV-1 CA hexamer | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.22 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
- Sample components
Sample components
-Entire : HIV-1 Capsid Protein Assembly
| Entire | Name: HIV-1 Capsid Protein Assembly |
|---|---|
| Components |
|
-Supramolecule #1: HIV-1 Capsid Protein Assembly
| Supramolecule | Name: HIV-1 Capsid Protein Assembly / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Human immunodeficiency virus type 1 (NEW YORK-5 ISOLATE) |
| Molecular weight | Theoretical: 24 kDa/nm |
-Macromolecule #1: Gag polyprotein
| Macromolecule | Name: Gag polyprotein / type: protein_or_peptide / ID: 1 / Number of copies: 6 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Human immunodeficiency virus type 1 (NEW YORK-5 ISOLATE) |
| Molecular weight | Theoretical: 24.654268 KDa |
| Recombinant expression | Organism:  |
| Sequence | String: PIVQNLQGQM VHQAISPRTL NAWVKVVEEK AFSPEVIPMF SALSEGATPQ DLNTMLNTVG GHQAAMQMLK ETINEEAAEW DRLHPVHAG PIAPGQMREP RGSDIAGTTS TLQEQIGWMT HNPPIPVGEI YKRWIILGLN KIVRMYSPTS ILDIRQGPKE P FRDYVDRF ...String: PIVQNLQGQM VHQAISPRTL NAWVKVVEEK AFSPEVIPMF SALSEGATPQ DLNTMLNTVG GHQAAMQMLK ETINEEAAEW DRLHPVHAG PIAPGQMREP RGSDIAGTTS TLQEQIGWMT HNPPIPVGEI YKRWIILGLN KIVRMYSPTS ILDIRQGPKE P FRDYVDRF YKTLRAEQAS QEVKNWMTET LLVQNANPDC KTILKALGPG ATLEEMMTAC QGV UniProtKB: Gag polyprotein |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | helical reconstruction |
| Aggregation state | helical array |
-Sample preparation
| Concentration | 2 mg/mL | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 8 Component:
| |||||||||
| Grid | Model: Quantifoil R2/1 / Material: COPPER / Mesh: 300 / Support film - Material: CARBON / Support film - topology: HOLEY ARRAY / Support film - Film thickness: 10 / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 25 sec. / Pretreatment - Atmosphere: AIR | |||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 295 K / Instrument: HOMEMADE PLUNGER Details: The assembled sample (1.5 microliter) was applied to the carbon side of a glow discharged perforated Quantifoil grid, followed by application of 3 microliter of low salt buffer (100 ...Details: The assembled sample (1.5 microliter) was applied to the carbon side of a glow discharged perforated Quantifoil grid, followed by application of 3 microliter of low salt buffer (100 milimolar NaCl, 50 milimolar Tris pH 8.0) on the back side of the grid, and blotting, from the back side, with a filter paper, before plunge-freezing in liquid ethane. |
- Electron microscopy
Electron microscopy
| Microscope | FEI POLARA 300 |
|---|---|
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: SUPER-RESOLUTION / Number real images: 523 / Average exposure time: 6.0 sec. / Average electron dose: 41.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 100.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.2 mm / Nominal defocus max: 2.2 µm / Nominal defocus min: 0.7000000000000001 µm / Nominal magnification: 31000 |
| Sample stage | Specimen holder model: SIDE ENTRY, EUCENTRIC / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Tecnai Polara / Image courtesy: FEI Company |