 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-7952: Caseinolytic protease (ClpP) from Staphylococcus aureus mutant - V7A -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-7952 | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Caseinolytic protease (ClpP) from Staphylococcus aureus mutant - V7A | |||||||||||||||||||||
 Map data Map data | primary map | |||||||||||||||||||||
 Sample Sample |
| |||||||||||||||||||||
 Keywords Keywords | Protease / HYDROLASE | |||||||||||||||||||||
| Function / homology |  Function and homology information Function and homology informationendopeptidase Clp / endopeptidase Clp complex / ATP-dependent peptidase activity / protein quality control for misfolded or incompletely synthesized proteins / ATPase binding / serine-type endopeptidase activity / identical protein binding / cytoplasm Similarity search - Function | |||||||||||||||||||||
| Biological species |  Staphylococcus aureus (strain Newman) (bacteria) / Staphylococcus aureus subsp. aureus str. Newman (bacteria) Staphylococcus aureus (strain Newman) (bacteria) / Staphylococcus aureus subsp. aureus str. Newman (bacteria) | |||||||||||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.7 Å | |||||||||||||||||||||
 Authors Authors | Ripstein ZA / Vahidi S / Kay LE / Rubinstein JL | |||||||||||||||||||||
| Funding support |  Canada, 6 items Canada, 6 items
| |||||||||||||||||||||
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2018 Title: Reversible inhibition of the ClpP protease via an N-terminal conformational switch. Authors: Siavash Vahidi / Zev A Ripstein / Massimiliano Bonomi / Tairan Yuwen / Mark F Mabanglo / Jordan B Juravsky / Kamran Rizzolo / Algirdas Velyvis / Walid A Houry / Michele Vendruscolo / John L ...Authors: Siavash Vahidi / Zev A Ripstein / Massimiliano Bonomi / Tairan Yuwen / Mark F Mabanglo / Jordan B Juravsky / Kamran Rizzolo / Algirdas Velyvis / Walid A Houry / Michele Vendruscolo / John L Rubinstein / Lewis E Kay /  Abstract: Protein homeostasis is critically important for cell viability. Key to this process is the refolding of misfolded or aggregated proteins by molecular chaperones or, alternatively, their degradation ...Protein homeostasis is critically important for cell viability. Key to this process is the refolding of misfolded or aggregated proteins by molecular chaperones or, alternatively, their degradation by proteases. In most prokaryotes and in chloroplasts and mitochondria, protein degradation is performed by the caseinolytic protease ClpP, a tetradecamer barrel-like proteolytic complex. Dysregulating ClpP function has shown promise in fighting antibiotic resistance and as a potential therapy for acute myeloid leukemia. Here we use methyl-transverse relaxation-optimized spectroscopy (TROSY)-based NMR, cryo-EM, biochemical assays, and molecular dynamics simulations to characterize the structural dynamics of ClpP from (SaClpP) in wild-type and mutant forms in an effort to discover conformational hotspots that regulate its function. Wild-type SaClpP was found exclusively in the active extended form, with the N-terminal domains of its component protomers in predominantly β-hairpin conformations that are less well-defined than other regions of the protein. A hydrophobic site was identified that, upon mutation, leads to unfolding of the N-terminal domains, loss of SaClpP activity, and formation of a previously unobserved split-ring conformation with a pair of 20-Å-wide pores in the side of the complex. The extended form of the structure and partial activity can be restored via binding of ADEP small-molecule activators. The observed structural plasticity of the N-terminal gates is shown to be a conserved feature through studies of and ClpP, suggesting a potential avenue for the development of molecules to allosterically modulate the function of ClpP. | |||||||||||||||||||||
| History |
|
- Structure visualization
Structure visualization
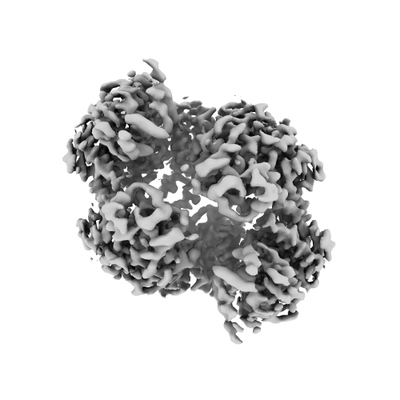
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_7952.map.gz | 3 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-7952-v30.xmlemd-7952.xml | 16.6 KB 16.6 KB | Display Display | EMDB header |
| Images |  emd_7952.png emd_7952.png | 48.3 KB | ||
| Filedesc metadata | emd-7952.cif.gz | 6 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-7952ftp://ftp.pdbj.org/pub/emdb/structures/EMD-7952 http://ftp.pdbj.org/pub/emdb/structures/EMD-7952ftp://ftp.pdbj.org/pub/emdb/structures/EMD-7952 | HTTPS FTP |
-Related structure data
| Related structure data |  6dkfMC  7950C  7951C M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_7952.map.gz / Format: CCP4 / Size: 27 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | primary map | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.06 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
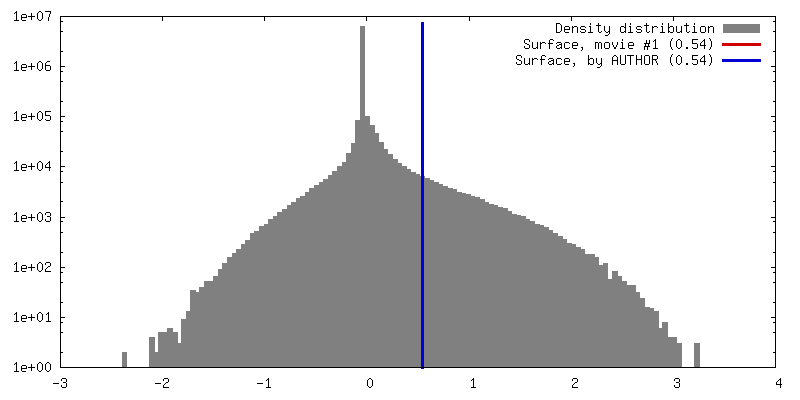
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)



















-Supplemental data
- Sample components
Sample components
-Entire : Caseinolytic protease from Staphylococcus aureus (V7A)
| Entire | Name: Caseinolytic protease from Staphylococcus aureus (V7A) |
|---|---|
| Components |
|
-Supramolecule #1: Caseinolytic protease from Staphylococcus aureus (V7A)
| Supramolecule | Name: Caseinolytic protease from Staphylococcus aureus (V7A) type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Staphylococcus aureus (strain Newman) (bacteria) / Strain: Newman |
| Molecular weight | Theoretical: 301 KDa |
-Macromolecule #1: ATP-dependent Clp protease proteolytic subunit
| Macromolecule | Name: ATP-dependent Clp protease proteolytic subunit / type: protein_or_peptide / ID: 1 / Number of copies: 14 / Enantiomer: LEVO / EC number: endopeptidase Clp |
|---|---|
| Source (natural) | Organism: Staphylococcus aureus subsp. aureus str. Newman (bacteria) Strain: Newman |
| Molecular weight | Theoretical: 21.508479 KDa |
| Recombinant expression | Organism: |
| Sequence | String: MNLIPTAIET TNRGERAYDI YSRLLKDRII MLGSQIDDNV ANSIVSQLLF LQAQDSEKDI YLYINSPGGS VTAGFAIYDT IQHIKPDVQ TICIGMAASM GSFLLAAGAK GKRFALPNAE VMIHQPLGGA QGQATEIEIA ANHILKTREK LNRILSERTG Q SIEKIQKD ...String: MNLIPTAIET TNRGERAYDI YSRLLKDRII MLGSQIDDNV ANSIVSQLLF LQAQDSEKDI YLYINSPGGS VTAGFAIYDT IQHIKPDVQ TICIGMAASM GSFLLAAGAK GKRFALPNAE VMIHQPLGGA QGQATEIEIA ANHILKTREK LNRILSERTG Q SIEKIQKD TDRDNFLTAE EAKEYGLIDE VMVPETK UniProtKB: ATP-dependent Clp protease proteolytic subunit |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 30 mg/mL | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7 Component:
| ||||||||||||||||||
| Grid | Support film - topology: HOLEY / Support film - Film thickness: 35 / Details: unspecified | ||||||||||||||||||
| Vitrification | Cryogen name: ETHANE-PROPANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK III / Details: Modified Vitrobot. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: FEI FALCON III (4k x 4k) / Detector mode: COUNTING / Digitization - Dimensions - Width: 4096 pixel / Digitization - Dimensions - Height: 4096 pixel / Digitization - Frames/image: 1-30 / Number grids imaged: 2 / Number real images: 1837 / Average exposure time: 60.0 sec. / Average electron dose: 43.0 e/Å2 / Details: movies were collected with 44 fractions |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 100.0 µm / Calibrated defocus max: 3.2 µm / Calibrated defocus min: 1.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal magnification: 75000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing
-Atomic model buiding 1
| Initial model |
| ||||||
|---|---|---|---|---|---|---|---|
| Refinement | Space: REAL / Protocol: OTHER | ||||||
| Output model | PDB-6dkf: |


