| Entry | Database: PDB / ID: 4use
|
|---|
| Title | Human STK10 (LOK) with SB-633825 |
|---|
 Components Components | SERINE/THREONINE-PROTEIN KINASE 10 |
|---|
 Keywords Keywords | TRANSFERASE |
|---|
| Function / homology |  Function and homology information Function and homology information
lymphocyte aggregation / regulation of lymphocyte migration / RHOB GTPase cycle / RHOC GTPase cycle / RHOA GTPase cycle / specific granule membrane / protein autophosphorylation / non-specific serine/threonine protein kinase / protein phosphorylation / protein serine kinase activity ...lymphocyte aggregation / regulation of lymphocyte migration / RHOB GTPase cycle / RHOC GTPase cycle / RHOA GTPase cycle / specific granule membrane / protein autophosphorylation / non-specific serine/threonine protein kinase / protein phosphorylation / protein serine kinase activity / protein serine/threonine kinase activity / Neutrophil degranulation / protein homodimerization activity / extracellular exosome / ATP binding / identical protein binding / plasma membrane / cytosol / cytoplasmSimilarity search - Function Serine/threonine-protein kinase 10, catalytic domain / Polo kinase kinase / : / Polo kinase kinase / Transferase(Phosphotransferase) domain 1 / Transferase(Phosphotransferase); domain 1 / Phosphorylase Kinase; domain 1 / Phosphorylase Kinase; domain 1 / Serine/threonine-protein kinase, active site / Serine/Threonine protein kinases active-site signature. ...Serine/threonine-protein kinase 10, catalytic domain / Polo kinase kinase / : / Polo kinase kinase / Transferase(Phosphotransferase) domain 1 / Transferase(Phosphotransferase); domain 1 / Phosphorylase Kinase; domain 1 / Phosphorylase Kinase; domain 1 / Serine/threonine-protein kinase, active site / Serine/Threonine protein kinases active-site signature. / Protein kinase domain / Serine/Threonine protein kinases, catalytic domain / Protein kinase, ATP binding site / Protein kinases ATP-binding region signature. / Protein kinase domain profile. / Protein kinase domain / Protein kinase-like domain superfamily / 2-Layer Sandwich / Orthogonal Bundle / Mainly Alpha / Alpha BetaSimilarity search - Domain/homology |
|---|
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.65 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.65 Å |
|---|
 Authors Authors | Elkins, J.M. / Salah, E. / Szklarz, M. / von Delft, F. / Canning, P. / Raynor, J. / Bountra, C. / Edwards, A.M. / Knapp, S. |
|---|
 Citation Citation | Journal: Nat.Biotechnol. / Year: 2016
Title: Comprehensive Characterization of the Published Kinase Inhibitor Set.
Authors: Elkins, J.M. / Fedele, V. / Szklarz, M. / Abdul Azeez, K.R. / Salah, E. / Mikolajczyk, J. / Romanov, S. / Sepetov, N. / Huang, X.P. / Roth, B.L. / Al Haj Zen, A. / Fourches, D. / Muratov, E. ...Authors: Elkins, J.M. / Fedele, V. / Szklarz, M. / Abdul Azeez, K.R. / Salah, E. / Mikolajczyk, J. / Romanov, S. / Sepetov, N. / Huang, X.P. / Roth, B.L. / Al Haj Zen, A. / Fourches, D. / Muratov, E. / Tropsha, A. / Morris, J. / Teicher, B.A. / Kunkel, M. / Polley, E. / Lackey, K.E. / Atkinson, F.L. / Overington, J.P. / Bamborough, P. / Moller, S. / Price, D.J. / Willson, T.M. / Drewry, D.H. / Knapp, S. / Zuercher, W.J. |
|---|
| History | | Deposition | Jul 7, 2014 | Deposition site: PDBE / Processing site: PDBE |
|---|
| Revision 1.0 | Jul 22, 2015 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Nov 4, 2015 | Group: Database references |
|---|
| Revision 1.2 | Jan 20, 2016 | Group: Database references |
|---|
| Revision 1.3 | Jan 24, 2018 | Group: Structure summary / Category: audit_author / Item: _audit_author.name |
|---|
|
|---|
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj
 Assembly
Assembly



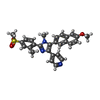
 Mass: 483.581 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C28H25N3O3S
Mass: 483.581 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C28H25N3O3S Mass: 18.015 Da / Num. of mol.: 16 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 16 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: I03 / Wavelength: 0.9763
/ Beamline: I03 / Wavelength: 0.9763  Processing
Processing