 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4j59 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Human Cyclophilin D Complexed with an Inhibitor | ||||||
 Components Components | Peptidyl-prolyl cis-trans isomerase F, mitochondrial | ||||||
 Keywords Keywords | Isomerase/Isomerase Inhibitor / Isomerase-Isomerase Inhibitor complex | ||||||
| Function / homology |  Function and homology information Function and homology informationmitochondrial outer membrane permeabilization involved in programmed cell death / regulation of mitochondrial membrane permeability involved in programmed necrotic cell death / mitochondrial permeability transition pore complex / cellular response to arsenic-containing substance / negative regulation of oxidative phosphorylation / regulation of mitochondrial membrane permeability / cyclosporin A binding / negative regulation of release of cytochrome c from mitochondria / negative regulation of intrinsic apoptotic signaling pathway / cellular response to calcium ion ...mitochondrial outer membrane permeabilization involved in programmed cell death / regulation of mitochondrial membrane permeability involved in programmed necrotic cell death / mitochondrial permeability transition pore complex / cellular response to arsenic-containing substance / negative regulation of oxidative phosphorylation / regulation of mitochondrial membrane permeability / cyclosporin A binding / negative regulation of release of cytochrome c from mitochondria / negative regulation of intrinsic apoptotic signaling pathway / cellular response to calcium ion / response to ischemia / enzyme inhibitor activity / peptidylprolyl isomerase / peptidyl-prolyl cis-trans isomerase activity / cellular response to hydrogen peroxide / protein folding / mitochondrial matrix / apoptotic process / negative regulation of apoptotic process / mitochondrion / membrane / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Isomorphous Replacement / Resolution: 1.92 Å X-RAY DIFFRACTION / SYNCHROTRON / Isomorphous Replacement / Resolution: 1.92 Å | ||||||
 Authors Authors | Gelin, M. / Colliandre, L. / Bessin, Y. / Guichou, J.F. | ||||||
 Citation Citation | Journal: Nat Commun / Year: 2016 Title: Fragment-based discovery of a new family of non-peptidic small-molecule cyclophilin inhibitors with potent antiviral activities. Authors: Ahmed-Belkacem, A. / Colliandre, L. / Ahnou, N. / Nevers, Q. / Gelin, M. / Bessin, Y. / Brillet, R. / Cala, O. / Douguet, D. / Bourguet, W. / Krimm, I. / Pawlotsky, J.M. / Guichou, J.F. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4j59.cif.gz | 77 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4j59.ent.gz | 56.6 KB | Display | PDB format |
| PDBx/mmJSON format | 4j59.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/j5/4j59ftp://data.pdbj.org/pub/pdb/validation_reports/j5/4j59 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3r49C  3r4gC  3r54C  3r56C  3r57C  3r59C  3rcfC  3rcgC  3rciC  3rckC  3rclC  3rd9C  3rdaC  3rdbC  3rddC  4j58C  4j5bC  4j5cC  4j5dC  4j5eC C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 17652.125 Da / Num. of mol.: 1 / Fragment: UNP residues 44-207 / Mutation: K175I Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: ppif, cyp3 / Production host:  |
|---|---|
| #2: Chemical | ChemComp-671 / 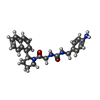  Mass: 402.489 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C24H26N4O2 Mass: 402.489 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C24H26N4O2 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 248 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 248 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.02 Å3/Da / Density % sol: 39.09 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 7.3 Details: 30% PEG4000, pH 7.3, vapor diffusion, hanging drop, temperature 291.0K |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID29 / Wavelength: 0.97908 Å / Beamline: ID29 / Wavelength: 0.97908 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Jun 10, 2010 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Asymmetric Laue 001 / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97908 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.92→40.357 Å / Num. all: 10847 / Num. obs: 10847 / % possible obs: 93.5 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 6.8 % / Biso Wilson estimate: 11.7 Å2 / Rmerge(I) obs: 0.109 / Rsym value: 0.109 / Net I/σ(I): 13.5 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: Isomorphous Replacement / Resolution: 1.92→40.353 Å / Occupancy max: 1 / Occupancy min: 0.31 / SU ML: 0.16 / σ(F): 1.39 / Phase error: 16.72 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 9.7856 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.92→40.353 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
