 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4gqj: Complex of a binuclear Ruthenium compound D,D-([mu-(11,11')-bi(dp... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4gqj | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Complex of a binuclear Ruthenium compound D,D-([mu-(11,11')-bi(dppz)-(1,10-phenanthroline)4-Ru2]4+) bound to d(CGTACG) | ||||||||||||||||||
 Components Components | DNA (5'-D(* Keywords KeywordsDNA / DNA recognition / DNA binsinf | Function / homology | Chem-RR2 / DNA |  Function and homology information Function and homology informationMethod |  X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2.951 Å X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2.951 Å  Authors AuthorsBoer, D.R. / Coll, M. |  CitationJournal: Angew.Chem.Int.Ed.Engl. / Year: 2014 CitationJournal: Angew.Chem.Int.Ed.Engl. / Year: 2014Title: Thread Insertion of a Bis(dipyridophenazine) Diruthenium Complex into the DNA Double Helix by the Extrusion of AT Base Pairs and Cross-Linking of DNA Duplexes. Authors: Boer, D.R. / Wu, L. / Lincoln, P. / Coll, M. History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4gqj.cif.gz | 20.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4gqj.ent.gz | 13 KB | Display | PDB format |
| PDBx/mmJSON format | 4gqj.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gq/4gqjftp://data.pdbj.org/pub/pdb/validation_reports/gq/4gqj | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |















-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: DNA chain | Mass: 1809.217 Da / Num. of mol.: 2 / Source method: obtained synthetically / Details: Acquired from biomers (Germany) #2: Chemical | ChemComp-RR2 / ( | 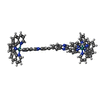  Mass: 1485.543 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C84H50N16Ru2 / Details: Synthesized by citation authors Mass: 1485.543 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C84H50N16Ru2 / Details: Synthesized by citation authors#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 16 / Source method: isolated from a natural source / Formula: H2O / Details: Synthesized by citation authors Mass: 18.015 Da / Num. of mol.: 16 / Source method: isolated from a natural source / Formula: H2O / Details: Synthesized by citation authors |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 7.18 Å3/Da / Density % sol: 82.87 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 8 Details: buffer: 0.2 M KCl 10 mM MgCl2 35% hexanediol 50 mM TRIS.HCl pH 8 drop composition: 0.5 ul 10 mM binuclear Ru compound 0.5 ul 10 mM [ssDNA] 5'-d(CGTACG)-3' 1 ul cryst. buffer 0.5 ul , VAPOR ...Details: buffer: 0.2 M KCl 10 mM MgCl2 35% hexanediol 50 mM TRIS.HCl pH 8 drop composition: 0.5 ul 10 mM binuclear Ru compound 0.5 ul 10 mM [ssDNA] 5'-d(CGTACG)-3' 1 ul cryst. buffer 0.5 ul , VAPOR DIFFUSION, SITTING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID29 / Wavelength: 1.07205 Å / Beamline: ID29 / Wavelength: 1.07205 Å |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Jun 14, 2009 |
| Radiation | Monochromator: channel-cut Si(111) crystal / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.07205 Å / Relative weight: 1 |
| Reflection | Resolution: 2.95→50 Å / Num. all: 2419 / Num. obs: 2419 / % possible obs: 97 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 4.4 % / Biso Wilson estimate: 73.4 Å2 / Rmerge(I) obs: 0.118 / Net I/σ(I): 10.3 |
| Reflection shell | Resolution: 2.95→3.11 Å / Redundancy: 4.6 % / Rmerge(I) obs: 0.62 / Mean I/σ(I) obs: 2.1 / Num. unique all: 343 / % possible all: 99 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD / Resolution: 2.951→38.205 Å / SU ML: 0.29 / σ(F): 1.36 / Phase error: 19.82 / Stereochemistry target values: ML
| |||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.72 Å / VDW probe radii: 1 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 19.848 Å2 / ksol: 0.272 e/Å3 | |||||||||||||||||||||||||
| Displacement parameters |
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.951→38.205 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| LS refinement shell | Highest resolution: 2.9511 Å
|
