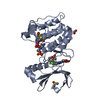
Entry Database : PDB / ID : 4bn4Title Structure of human SIRT3 in complex with ADP-ribose NAD-DEPENDENT PROTEIN DEACETYLASE SIRTUIN-3, MITOCHONDRIAL Keywords / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species HOMO SAPIENS (human)Method / / / Resolution : 1.3 Å Authors Nguyen, G.T.T. / Schaefer, S. / Gertz, M. / Weyand, M. / Steegborn, C. Journal : Acta Crystallogr.,Sect.D / Year : 2013Title : Structures of Human Sirtuin 3 Complexes with Adp-Ribose and with Carba-Nad+ and Srt1720: Binding Details and Inhibition MechanismAuthors : Nguyen, G.T.T. / Schaefer, S. / Gertz, M. / Weyand, M. / Steegborn, C. History Deposition May 13, 2013 Deposition site / Processing site Revision 1.0 Jun 26, 2013 Provider / Type Revision 1.1 Jul 24, 2013 Group / OtherRevision 1.2 Jul 31, 2013 Group Revision 1.3 Aug 7, 2013 Group / OtherRevision 1.4 Aug 14, 2013 Group / OtherRevision 1.5 Aug 28, 2013 Group / OtherRevision 1.6 Dec 20, 2023 Group Data collection / Database references ... Data collection / Database references / Derived calculations / Other / Refinement description / Structure summary Category chem_comp / chem_comp_atom ... chem_comp / chem_comp_atom / chem_comp_bond / database_2 / pdbx_database_status / pdbx_initial_refinement_model / pdbx_struct_conn_angle / struct_conn / struct_site Item _chem_comp.pdbx_synonyms / _database_2.pdbx_DOI ... _chem_comp.pdbx_synonyms / _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_database_status.status_code_sf / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr2_auth_comp_id / _pdbx_struct_conn_angle.ptnr2_auth_seq_id / _pdbx_struct_conn_angle.ptnr2_label_asym_id / _pdbx_struct_conn_angle.ptnr2_label_atom_id / _pdbx_struct_conn_angle.ptnr2_label_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.value / _struct_conn.pdbx_dist_value / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_conn.ptnr2_label_seq_id / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id
Show all Show less
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information HOMO SAPIENS (human)
HOMO SAPIENS (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj











 Assembly
Assembly





 Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na
Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 559.316 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H23N5O14P2
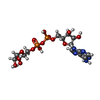
Mass: 559.316 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H23N5O14P2 Mass: 296.314 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H24O8
Mass: 296.314 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H24O8 Sample preparation
Sample preparation / Beamline: 14.1 / Wavelength: 0.91841
/ Beamline: 14.1 / Wavelength: 0.91841  Processing
Processing