Mass: 35940.777 Da / Num. of mol.: 2 / Fragment: RESIDUES 33-243,272-368 / Mutation: YES Source method: isolated from a genetically manipulated source Details: RESIDUES 3-32 AT THE N-TERMINUS AND RESIDUES 244-271 OF THE THIRD INTRACELLULAR LOOP WERE DELETED FROM THE CONSTRUCT. THE CONSTRUCT WAS TRUNCATED AFTER RESIDUE 367 AND A HEXAHIS TAG ADDED. Source: (gene. exp.) MELEAGRIS GALLOPAVO (turkey) / Cell: ERYTHROCYTE / Plasmid: PBACPAK8 / Cell line (production host): High Five / Production host: TRICHOPLUSIA NI (cabbage looper) / References: UniProt: P07700
Mass: 18.015 Da / Num. of mol.: 35 / Source method: isolated from a natural source / Formula: H2O
-
Details
Has protein modification
Y
Sequence details
THE FOLLOWING MUTATIONS WERE MADE TO IMPROVE THERMOSTABILITY R68S,M90V,Y227A,A282L,F327A,F338M THE ...THE FOLLOWING MUTATIONS WERE MADE TO IMPROVE THERMOSTABILITY R68S,M90V,Y227A,A282L,F327A,F338M THE FOLLOWING MUTATIONS WERE MADE TO IMPROVE EXPRESSION AND HELP CRYSTALLISATION C116L, C358A
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 3.63 Å3/Da / Density % sol: 66.14 % / Description: NONE
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj







 Assembly
Assembly


 TRICHOPLUSIA NI (cabbage looper) / References: UniProt: P07700
TRICHOPLUSIA NI (cabbage looper) / References: UniProt: P07700

 Mass: 22.990 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Na
Mass: 22.990 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Na Mass: 486.726 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C31H50O4
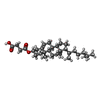
Mass: 486.726 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C31H50O4 Mass: 227.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H17N3
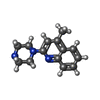
Mass: 227.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H17N3 Mass: 379.489 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C18H37NO7 / Comment: detergent*YM
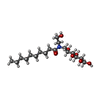
Mass: 379.489 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C18H37NO7 / Comment: detergent*YM Sample preparation
Sample preparation / Beamline: I24 / Wavelength: 0.9778
/ Beamline: I24 / Wavelength: 0.9778  Processing
Processing