 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3vjm: Crystal structure of human depiptidyl peptidase IV (DPP-4) in com... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3vjm | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of human depiptidyl peptidase IV (DPP-4) in complex with a prolylthiazolidine inhibitor #1 | |||||||||
 Components Components | Dipeptidyl peptidase 4 | |||||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / ALPHA/BETA / BETA-PROPELLER / AMINOPEPTIDASE / SERINE PROTEASE / SIGNAL-ANCHOR / TRANSMEMBRANE / DIABETES / GLYCOPROTEIN / CELL MEMBRANE / HYDROLASE-HYDROLASE INHIBITOR complex | |||||||||
| Function / homology |  Function and homology information Function and homology informationglucagon processing / negative regulation of neutrophil chemotaxis / regulation of cell-cell adhesion mediated by integrin / Synthesis, secretion, and inactivation of Glucose-dependent Insulinotropic Polypeptide (GIP) / dipeptidyl-peptidase IV / negative regulation of extracellular matrix disassembly / chemorepellent activity / intercellular canaliculus / psychomotor behavior / dipeptidyl-peptidase activity ...glucagon processing / negative regulation of neutrophil chemotaxis / regulation of cell-cell adhesion mediated by integrin / Synthesis, secretion, and inactivation of Glucose-dependent Insulinotropic Polypeptide (GIP) / dipeptidyl-peptidase IV / negative regulation of extracellular matrix disassembly / chemorepellent activity / intercellular canaliculus / psychomotor behavior / dipeptidyl-peptidase activity / peptide hormone processing / lamellipodium membrane / locomotory exploration behavior / aminopeptidase activity / endocytic vesicle / endothelial cell migration / behavioral fear response / T cell costimulation / receptor-mediated endocytosis of virus by host cell / serine-type peptidase activity / T cell activation / Synthesis, secretion, and inactivation of Glucagon-like Peptide-1 (GLP-1) / lamellipodium / virus receptor activity / protease binding / membrane fusion / response to hypoxia / receptor-mediated virion attachment to host cell / cell adhesion / apical plasma membrane / membrane raft / signaling receptor binding / serine-type endopeptidase activity / lysosomal membrane / focal adhesion / positive regulation of cell population proliferation / symbiont entry into host cell / cell surface / protein homodimerization activity / proteolysis / extracellular exosome / extracellular region / membrane / identical protein binding / plasma membrane Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å | |||||||||
 Authors Authors | Akahoshi, F. / Kishida, H. / Miyaguchi, I. / Yoshida, T. / Ishii, S. | |||||||||
 Citation Citation | Journal: Bioorg.Med.Chem. / Year: 2012 Title: Fused bicyclic heteroarylpiperazine-substituted l-prolylthiazolidines as highly potent DPP-4 inhibitors lacking the electrophilic nitrile group Authors: Yoshida, T. / Akahoshi, F. / Sakashita, H. / Sonda, S. / Takeuchi, M. / Tanaka, Y. / Nabeno, M. / Kishida, H. / Miyaguchi, I. / Hayashi, Y. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3vjm.cif.gz | 332.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3vjm.ent.gz | 266.5 KB | Display | PDB format |
| PDBx/mmJSON format | 3vjm.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/vj/3vjmftp://data.pdbj.org/pub/pdb/validation_reports/vj/3vjm | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1j2eS S: Starting model for refinement |
|---|---|
| Similar structure data |






















-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 85880.031 Da / Num. of mol.: 2 / Fragment: UNP residues 33-766 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: DPP4, ADCP2, CD26 / Plasmid: pPSC8 / Cell line (production host): expresSF+ / Production host:   Spodoptera frugiperda (fall armyworm) / References: UniProt: P27487, dipeptidyl-peptidase IV Spodoptera frugiperda (fall armyworm) / References: UniProt: P27487, dipeptidyl-peptidase IV#2: Polysaccharide | 2-acetamido-2-deoxy-beta-D-glucopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose Source method: isolated from a genetically manipulated source #3: Chemical | 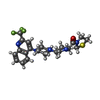  Mass: 465.535 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C22H26F3N5OS Mass: 465.535 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C22H26F3N5OS#4: Sugar | ChemComp-NAG /   Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 7 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 7Source method: isolated from a genetically manipulated source Formula: C8H15NO6 #5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 987 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 987 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.97 Å3/Da / Density % sol: 58.54 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 8.8 Details: 20% PEG 4000, 0.18M sodium acetate, 0.18M glycine-sodium hydroxide, pH 8.8, VAPOR DIFFUSION, SITTING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Photon Factory  / Beamline: BL-5A / Wavelength: 1 Å / Beamline: BL-5A / Wavelength: 1 Å |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Nov 28, 2006 / Details: mirrors |
| Radiation | Monochromator: Si 111 / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→31.342 Å / Num. all: 119422 / Num. obs: 109648 / % possible obs: 91 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 4.8 % / Biso Wilson estimate: 28.1 Å2 / Rmerge(I) obs: 0.067 / Net I/σ(I): 11.7 |
| Reflection shell | Resolution: 2.1→2.18 Å / Redundancy: 4.5 % / Rmerge(I) obs: 0.399 / Num. unique all: 10763 / % possible all: 91.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1J2E Resolution: 2.1→30 Å / Cor.coef. Fo:Fc: 0.952 / Cor.coef. Fo:Fc free: 0.932 / SU B: 4.172 / SU ML: 0.112 / Isotropic thermal model: isotropic / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R Free: 0.176 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 28.616 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→30 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.1→2.154 Å / Total num. of bins used: 20
|
