 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3vev | ||||||
|---|---|---|---|---|---|---|---|
| Title | Glucokinase in complex with an activator and glucose | ||||||
 Components Components | Glucokinase | ||||||
 Keywords Keywords | TRANSFERASE/TRANSFERASE ACTIVATOR / catalysis reaction / TRANSFERASE / TRANSFERASE-TRANSFERASE ACTIVATOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationDefective GCK causes maturity-onset diabetes of the young 2 (MODY2) / glucose sensor activity / mannokinase activity / regulation of potassium ion transport / hexokinase / fructokinase activity / carbohydrate phosphorylation / glucose catabolic process / glucokinase activity / glucose 6-phosphate metabolic process ...Defective GCK causes maturity-onset diabetes of the young 2 (MODY2) / glucose sensor activity / mannokinase activity / regulation of potassium ion transport / hexokinase / fructokinase activity / carbohydrate phosphorylation / glucose catabolic process / glucokinase activity / glucose 6-phosphate metabolic process / NADP metabolic process / Regulation of Glucokinase by Glucokinase Regulatory Protein / Defective TPR may confer susceptibility towards thyroid papillary carcinoma (TPC) / cellular response to leptin stimulus / D-glucose binding / canonical glycolysis / calcium ion import / Glycolysis / regulation of glycolytic process / intracellular glucose homeostasis / Regulation of gene expression in beta cells / positive regulation of glycogen biosynthetic process / negative regulation of gluconeogenesis / FOXO-mediated transcription of oxidative stress, metabolic and neuronal genes / response to glucose / regulation of insulin secretion / glycolytic process / positive regulation of insulin secretion / cellular response to insulin stimulus / glucose metabolic process / glucose homeostasis / mitochondrion / nucleoplasm / ATP binding / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å | ||||||
 Authors Authors | Liu, S. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2012 Title: Insights into Mechanism of Glucokinase Activation: OBSERVATION OF MULTIPLE DISTINCT PROTEIN CONFORMATIONS. Authors: Liu, S. / Ammirati, M.J. / Song, X. / Knafels, J.D. / Zhang, J. / Greasley, S.E. / Pfefferkorn, J.A. / Qiu, X. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3vev.cif.gz | 201.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3vev.ent.gz | 158.1 KB | Display | PDB format |
| PDBx/mmJSON format | 3vev.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 3vev_validation.pdf.gz | 732.1 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 3vev_full_validation.pdf.gz | 735.7 KB | Display | |
| Data in XML | 3vev_validation.xml.gz | 21.4 KB | Display | |
| Data in CIF | 3vev_validation.cif.gz | 32.5 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ve/3vevftp://data.pdbj.org/pub/pdb/validation_reports/ve/3vev | HTTPS FTP |
-Related structure data
| Related structure data |  3veyC  3vf6C  4dchC  4dhyC  3f9mS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj











- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 52996.188 Da / Num. of mol.: 1 / Fragment: UNP residues 12-465 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: GCK / Production host:  |
|---|---|
| #2: Sugar | ChemComp-GLC / 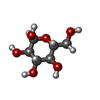  Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 1 Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Formula: C6H12O6 |
| #3: Chemical | ChemComp-0H4 / (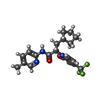  Mass: 393.403 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H22F3N3O2 Mass: 393.403 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H22F3N3O2 |
| #4: Chemical | ChemComp-NA /   Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 362 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 362 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.24 Å3/Da / Density % sol: 45.17 % |
|---|---|
| Crystal grow | Method: vapor diffusion, hanging drop / pH: 7 Details: Protein was in 25 mM HEPES pH 7.0, 0.5 mM TCEP, 0.05 M NaCl, 40 mM glucose at 20 mg/mL and 1 mM a GKA. Crystallization drops in a 1:1 ratio were set up over wells containing 0.1 M Tris HCl, ...Details: Protein was in 25 mM HEPES pH 7.0, 0.5 mM TCEP, 0.05 M NaCl, 40 mM glucose at 20 mg/mL and 1 mM a GKA. Crystallization drops in a 1:1 ratio were set up over wells containing 0.1 M Tris HCl, pH 7.0, 80-200 mM glucose, and 19-26% PEG-4000, VAPOR DIFFUSION, HANGING DROP |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 17-ID / Wavelength: 1 Å / Beamline: 17-ID / Wavelength: 1 Å |
| Detector | Type: PSI PILATUS 6M / Detector: PIXEL / Date: Jun 17, 2011 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.77→86 Å / Num. obs: 47167 / % possible obs: 99.3 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 2 / Redundancy: 6.7 % / Biso Wilson estimate: 22.57 Å2 / Rmerge(I) obs: 0.056 / Net I/σ(I): 22.1 |
- Processing
Processing
| Software | Name: BUSTER / Version: 2.11.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3F9M Resolution: 1.8→18.14 Å / Cor.coef. Fo:Fc: 0.9498 / Cor.coef. Fo:Fc free: 0.9423 / SU R Cruickshank DPI: 0.115 / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 28.01 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.185 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→18.14 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.8→1.85 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: -27.5225 Å / Origin y: -0.6825 Å / Origin z: 9.664 Å
|
