 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3v8w: Crystal Structure of Interleukin-2 Inducible T-cell Kinase Itk Ca... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3v8w | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Interleukin-2 Inducible T-cell Kinase Itk Catalytic Domain with Thienopyrazolylindole Inhibitor 469 | ||||||
 Components Components | Tyrosine-protein kinase ITK/TSK | ||||||
 Keywords Keywords | TRANSFERASE/TRANSFERASE INHIBITOR / kinase / TRANSFERASE-TRANSFERASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationNK T cell differentiation / gamma-delta T cell activation / Generation of second messenger molecules / cellular defense response / FCERI mediated Ca+2 mobilization / T cell activation / positive regulation of cytokine production / B cell receptor signaling pathway / non-specific protein-tyrosine kinase / non-membrane spanning protein tyrosine kinase activity ...NK T cell differentiation / gamma-delta T cell activation / Generation of second messenger molecules / cellular defense response / FCERI mediated Ca+2 mobilization / T cell activation / positive regulation of cytokine production / B cell receptor signaling pathway / non-specific protein-tyrosine kinase / non-membrane spanning protein tyrosine kinase activity / cell-cell junction / T cell receptor signaling pathway / adaptive immune response / intracellular signal transduction / signal transduction / zinc ion binding / ATP binding / nucleus / plasma membrane / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.27 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.27 Å | ||||||
 Authors Authors | McLean, L.R. / Zhang, Y. | ||||||
 Citation Citation | Journal: Bioorg.Med.Chem.Lett. / Year: 2012 Title: X-ray crystallographic structure-based design of selective thienopyrazole inhibitors for interleukin-2-inducible tyrosine kinase. Authors: McLean, L.R. / Zhang, Y. / Zaidi, N. / Bi, X. / Wang, R. / Dharanipragada, R. / Jurcak, J.G. / Gillespy, T.A. / Zhao, Z. / Musick, K.Y. / Choi, Y.M. / Barrague, M. / Peppard, J. / Smicker, M. ...Authors: McLean, L.R. / Zhang, Y. / Zaidi, N. / Bi, X. / Wang, R. / Dharanipragada, R. / Jurcak, J.G. / Gillespy, T.A. / Zhao, Z. / Musick, K.Y. / Choi, Y.M. / Barrague, M. / Peppard, J. / Smicker, M. / Duguid, M. / Parkar, A. / Fordham, J. / Kominos, D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3v8w.cif.gz | 108.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3v8w.ent.gz | 81.8 KB | Display | PDB format |
| PDBx/mmJSON format | 3v8w.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/v8/3v8wftp://data.pdbj.org/pub/pdb/validation_reports/v8/3v8w | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3v5jC  3v5lC  3v8tC  3vf8C  3vf9C  1sm2S C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj











- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 30248.551 Da / Num. of mol.: 2 / Fragment: unp residues 357-620 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: ITK, EMT, LYK / Production host:   Spodoptera frugiperda (fall armyworm) Spodoptera frugiperda (fall armyworm)References: UniProt: Q08881, non-specific protein-tyrosine kinase #2: Chemical | 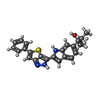  Mass: 401.524 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C24H23N3OS Mass: 401.524 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C24H23N3OS#3: Chemical | ChemComp-SO4 / |   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 49 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 49 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.73 Å3/Da / Density % sol: 54.9 % |
|---|---|
| Crystal grow | Temperature: 294 K / Method: vapor diffusion / pH: 5.8 Details: 0.8M Ammonium-sulfate, 0.1 M Na-citrate, 0.2 M Mg-acetate, 10 mM DTT, pH 5.8, VAPOR DIFFUSION, temperature 294K |
-Data collection
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH3R / Wavelength: 1.5418 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Detector | Type: RIGAKU RAXIS IV++ / Detector: IMAGE PLATE / Date: Dec 16, 2005 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.26→50 Å / Num. obs: 30265 / % possible obs: 99.6 % / Redundancy: 4 % / Rmerge(I) obs: 0.185 / Χ2: 2.266 / Net I/σ(I): 5.9 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1sm2 Resolution: 2.27→38.5 Å / Occupancy max: 1 / Occupancy min: 1 / SU R Cruickshank DPI: 0.285 / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 124.09 Å2 / Biso mean: 52.9913 Å2 / Biso min: 24.32 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.474 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.27→38.5 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.27→2.35 Å / Total num. of bins used: 15
|
