 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3sml: Crystal structure of human 14-3-3 sigma C38N/N166H in complex wit... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3sml | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of human 14-3-3 sigma C38N/N166H in complex with TASK-3 peptide and stabilizer Fusicoccin A aglycone | ||||||
 Components Components |
| ||||||
 Keywords Keywords | PEPTIDE BINDING PROTEIN / Helical protein / Phosphoprotein / Adapter protein / Nucleus | ||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation of aldosterone secretion / regulation of action potential firing rate / TWIK-releated acid-sensitive K+ channel (TASK) / Phase 4 - resting membrane potential / potassium ion leak channel activity / regulation of resting membrane potential / cellular response to acidic pH / outward rectifier potassium channel activity / regulation of epidermal cell division / protein kinase C inhibitor activity ...negative regulation of aldosterone secretion / regulation of action potential firing rate / TWIK-releated acid-sensitive K+ channel (TASK) / Phase 4 - resting membrane potential / potassium ion leak channel activity / regulation of resting membrane potential / cellular response to acidic pH / outward rectifier potassium channel activity / regulation of epidermal cell division / protein kinase C inhibitor activity / sodium channel activity / positive regulation of epidermal cell differentiation / keratinocyte development / keratinization / potassium ion import across plasma membrane / regulation of cell-cell adhesion / cAMP/PKA signal transduction / Regulation of localization of FOXO transcription factors / keratinocyte proliferation / phosphoserine residue binding / potassium channel activity / Activation of BAD and translocation to mitochondria / negative regulation of keratinocyte proliferation / establishment of skin barrier / negative regulation of protein localization to plasma membrane / Chk1/Chk2(Cds1) mediated inactivation of Cyclin B:Cdk1 complex / SARS-CoV-2 targets host intracellular signalling and regulatory pathways / negative regulation of protein kinase activity / negative regulation of stem cell proliferation / SARS-CoV-1 targets host intracellular signalling and regulatory pathways / RHO GTPases activate PKNs / positive regulation of protein localization / visual perception / positive regulation of cell adhesion / protein sequestering activity / negative regulation of innate immune response / protein export from nucleus / TP53 Regulates Transcription of Genes Involved in G2 Cell Cycle Arrest / release of cytochrome c from mitochondria / positive regulation of protein export from nucleus / stem cell proliferation / Translocation of SLC2A4 (GLUT4) to the plasma membrane / TP53 Regulates Metabolic Genes / potassium ion transport / intrinsic apoptotic signaling pathway in response to DNA damage / synaptic vesicle / intracellular protein localization / regulation of protein localization / positive regulation of cell growth / mitochondrial inner membrane / regulation of cell cycle / cadherin binding / protein heterodimerization activity / dendrite / protein kinase binding / negative regulation of transcription by RNA polymerase II / signal transduction / extracellular space / extracellular exosome / metal ion binding / identical protein binding / nucleus / plasma membrane / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.9 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | ||||||
 Authors Authors | Anders, C. / Schumacher, B. / Ottmann, C. | ||||||
 Citation Citation | Journal: Chem. Biol. / Year: 2013 Title: A semisynthetic fusicoccane stabilizes a protein-protein interaction and enhances the expression of K+ channels at the cell surface. Authors: Anders, C. / Higuchi, Y. / Koschinsky, K. / Bartel, M. / Schumacher, B. / Thiel, P. / Nitta, H. / Preisig-Muller, R. / Schlichthorl, G. / Renigunta, V. / Ohkanda, J. / Daut, J. / Kato, N. / Ottmann, C. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3sml.cif.gz | 75.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3sml.ent.gz | 55.1 KB | Display | PDB format |
| PDBx/mmJSON format | 3sml.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 3sml_validation.pdf.gz | 771.2 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 3sml_full_validation.pdf.gz | 774.9 KB | Display | |
| Data in XML | 3sml_validation.xml.gz | 14.8 KB | Display | |
| Data in CIF | 3sml_validation.cif.gz | 22 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/sm/3smlftp://data.pdbj.org/pub/pdb/validation_reports/sm/3sml | HTTPS FTP |
-Related structure data
| Related structure data |  3p1nC  3p1oC  3p1pC  3p1qC  3p1rC  3p1sC  3smkC  3smmC  3smnC  3smoC  3sp5C  3sprC  3ux0C  4fr3C  3lw1S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj












- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 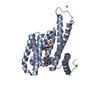
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
-Protein / Protein/peptide , 2 types, 2 molecules AP
| #1: Protein | Mass: 26577.920 Da / Num. of mol.: 1 / Mutation: C38N N166H Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: HME1, SFN / Plasmid: pPROEX HTB / Production host:  |
|---|---|
| #2: Protein/peptide | Mass: 727.770 Da / Num. of mol.: 1 / Source method: obtained synthetically / Details: This sequence occurs naturally in humans. / Source: (synth.) Homo sapiens (human) / References: UniProt: Q9NPC2*PLUS |
-Non-polymers , 4 types, 242 molecules 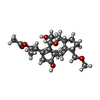



| #3: Chemical | ChemComp-FW1 /  Mass: 408.528 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H36O6 Mass: 408.528 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H36O6 | ||
|---|---|---|---|
| #4: Chemical | ChemComp-CL /  Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl | ||
| #5: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 236 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.64 Å3/Da / Density % sol: 53.45 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 7.4 Details: 0.095M HEPES Na-Salt pH7.4, 25.6% PEG 400, 0.19M CaCl2, 5% Glycerol, VAPOR DIFFUSION, HANGING DROP, temperature 277K |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: BRUKER AXS MICROSTAR / Wavelength: 1.5418 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MAR scanner 345 mm plate / Detector: IMAGE PLATE / Date: Jan 24, 2011 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.9→19.6 Å / Num. all: 23201 / Num. obs: 23128 / % possible obs: 99.7 % / Observed criterion σ(F): -3 / Observed criterion σ(I): -3 / Biso Wilson estimate: 12.319 Å2 / Rmerge(I) obs: 0.063 / Net I/σ(I): 24.08 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB Entry 3LW1 Resolution: 1.9→19.55 Å / Cor.coef. Fo:Fc: 0.95 / Cor.coef. Fo:Fc free: 0.918 / Occupancy max: 1 / Occupancy min: 0.3 / SU B: 2.464 / SU ML: 0.076 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.138 / ESU R Free: 0.132 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 48.17 Å2 / Biso mean: 12.3194 Å2 / Biso min: 2 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→19.55 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.9→1.949 Å / Total num. of bins used: 20
|
