 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3op5 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Human vaccinia-related kinase 1 | ||||||
 Components Components | Serine/threonine-protein kinase VRK1 | ||||||
 Keywords Keywords | TRANSFERASE / Adenosine Triphosphate / Amino Acid Sequence / Binding Sites / Catalytic Domain / Models / Molecular / Molecular Sequence Data / Phosphotransferases / Protein Conformation / Protein Folding / Surface Entropy Reduction / Structural Genomics / Structural Genomics Consortium / SGC | ||||||
| Function / homology |  Function and homology information Function and homology informationhistone H2AX kinase activity / Cajal body organization / Golgi disassembly / histone H3T3 kinase activity / Nuclear Envelope Breakdown / positive regulation of protein localization to chromatin / regulation of neuron migration / mitotic nuclear membrane disassembly / histone H3S10 kinase activity / Golgi stack ...histone H2AX kinase activity / Cajal body organization / Golgi disassembly / histone H3T3 kinase activity / Nuclear Envelope Breakdown / positive regulation of protein localization to chromatin / regulation of neuron migration / mitotic nuclear membrane disassembly / histone H3S10 kinase activity / Golgi stack / Initiation of Nuclear Envelope (NE) Reformation / Cajal body / nucleosomal DNA binding / neuron projection development / kinase activity / protein autophosphorylation / histone binding / protein phosphorylation / protein kinase activity / non-specific serine/threonine protein kinase / chromatin remodeling / protein serine kinase activity / cell division / protein serine/threonine kinase activity / DNA damage response / protein kinase binding / chromatin / signal transduction / nucleoplasm / ATP binding / nucleus / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å | ||||||
 Authors Authors | Allerston, C.K. / Uttarkar, S. / Savitsky, P. / Elkins, J.M. / Filippakopoulos, P. / Krojer, T. / Rellos, P. / Fedorov, O. / Eswaran, J. / Brenner, B. ...Allerston, C.K. / Uttarkar, S. / Savitsky, P. / Elkins, J.M. / Filippakopoulos, P. / Krojer, T. / Rellos, P. / Fedorov, O. / Eswaran, J. / Brenner, B. / Keates, T. / Das, S. / King, O. / Chalk, R. / Berridge, G. / von Delft, F. / Gileadi, O. / Arrowsmith, C.H. / Edwards, A.M. / Weigelt, J. / Bountra, C. / Knapp, S. / Structural Genomics Consortium (SGC) | ||||||
 Citation Citation | Journal: Sci Rep / Year: 2017 Title: Structural characterization of human Vaccinia-Related Kinases (VRK) bound to small-molecule inhibitors identifies different P-loop conformations. Authors: Counago, R.M. / Allerston, C.K. / Savitsky, P. / Azevedo, H. / Godoi, P.H. / Wells, C.I. / Mascarello, A. / de Souza Gama, F.H. / Massirer, K.B. / Zuercher, W.J. / Guimaraes, C.R.W. / Gileadi, O. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3op5.cif.gz | 523.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3op5.ent.gz | 428 KB | Display | PDB format |
| PDBx/mmJSON format | 3op5.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/op/3op5ftp://data.pdbj.org/pub/pdb/validation_reports/op/3op5 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5ukfC  5uu1C  5uvfC  1ckjS  2jiiS  2v62S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 41138.125 Da / Num. of mol.: 4 / Fragment: KINASE DOMAIN, RESIDUES 3-369 Mutation: K34A, K35A, E36A, E212A, K214A, E215A, E292A, K293A, K295A, K359A, K360A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: VRK1 / Plasmid: pNIC28-Bsa4 / Production host:  References: UniProt: Q99986, non-specific serine/threonine protein kinase #2: Chemical | ChemComp-REB / [ 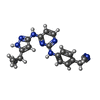  Mass: 331.374 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C18H17N7 Mass: 331.374 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C18H17N7#3: Chemical | ChemComp-EDO /   Mass: 62.068 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C2H6O2#4: Chemical |   Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 900 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 900 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.64 Å3/Da / Density % sol: 53.33 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 7 Details: 0.2m K/Na(tartrate), 20% PEG 3350, pH 7.0, VAPOR DIFFUSION, SITTING DROP, temperature 277K |
-Data collection
| Diffraction | Mean temperature: 200 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I02 / Wavelength: 0.9795 Å / Beamline: I02 / Wavelength: 0.9795 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MARMOSAIC 225 mm CCD / Detector: CCD / Date: Jun 30, 2010 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Si (111) double crystal / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.9795 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Redundancy: 3.5 % / Av σ(I) over netI: 6.2 / Number: 240388 / Rsym value: 0.113 / D res high: 2.4 Å / D res low: 46.338 Å / Num. obs: 67821 / % possible obs: 98.9 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Diffraction reflection shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.4→46.338 Å / Num. all: 67821 / Num. obs: 67821 / % possible obs: 98.9 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 3.5 % / Biso Wilson estimate: 47.65 Å2 / Rsym value: 0.113 / Net I/σ(I): 7.8 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: ENSEMBL OF PDB ENTRIES 2V62, 2JII and 1CKJ Resolution: 2.4→46.338 Å / Cor.coef. Fo:Fc: 0.9412 / Cor.coef. Fo:Fc free: 0.9127 / Occupancy max: 1 / Occupancy min: 0.05 / SU B: 7.378 / SU ML: 0.169 / SU R Cruickshank DPI: 0.31 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.31 / ESU R Free: 0.242 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 46.15 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.285 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→46.338 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.4→2.46 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
