 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2y3p: Crystal structure of N-terminal domain of GyrA with the antibioti... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2y3p | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of N-terminal domain of GyrA with the antibiotic simocyclinone D8 | |||||||||
 Components Components | DNA GYRASE SUBUNIT A | |||||||||
 Keywords Keywords | ISOMERASE / AMINOCOUMARIN ANTIBIOTIC | |||||||||
| Function / homology |  Function and homology information Function and homology informationDNA topoisomerase type II (double strand cut, ATP-hydrolyzing) complex / DNA negative supercoiling activity / DNA topoisomerase (ATP-hydrolysing) / DNA topological change / DNA-templated DNA replication / chromosome / response to antibiotic / DNA binding / ATP binding / cytoplasm Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.62 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.62 Å | |||||||||
 Authors Authors | Edwards, M.J. / Flatman, R.H. / Mitchenall, L.A. / Stevenson, C.E.M. / Le, T.B.K. / Clarke, T.A. / McKay, A.R. / Fiedler, H.-P. / Buttner, M.J. / Lawson, D.M. / Maxwell, A. | |||||||||
 Citation Citation | Journal: Science / Year: 2009 Title: A Crystal Structure of the Bifunctional Antibiotic Simocyclinone D8, Bound to DNA Gyrase. Authors: Edwards, M.J. / Flatman, R.H. / Mitchenall, L.A. / Stevenson, C.E.M. / Le, T.B.K. / Clarke, T.A. / Mckay, A.R. / Fiedler, H.-P. / Buttner, M.J. / Lawson, D.M. / Maxwell, A. #1: Journal: Acta Crystallogr.,Sect.F / Year: 2009 Title: Crystallization and Preliminary X-Ray Analysis of a Complex Formed between the Antibiotic Simocyclinone D8 and the DNA Breakage-Reunion Domain of Escherichia Coli DNA Gyrase. Authors: Edwards, M.J. / Flatman, R.H. / Mitchenall, L.A. / Stevenson, C.E.M. / Maxwell, A. / Lawson, D.M. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2y3p.cif.gz | 397.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2y3p.ent.gz | 325.4 KB | Display | PDB format |
| PDBx/mmJSON format | 2y3p.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/y3/2y3pftp://data.pdbj.org/pub/pdb/validation_reports/y3/2y3p | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1ab4S S: Starting model for refinement |
|---|---|
| Similar structure data |






-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (0.158, 0.983, -0.098), Vector: |
-Components
| #1: Protein | Mass: 58623.027 Da / Num. of mol.: 2 / Fragment: N-TERMINAL 59KDA DOMAIN, RESIDUES 2-523 Source method: isolated from a genetically manipulated source Details: COMPRISES RESIDUES 2-523 OF WILD TYPE SEQUENCE / Source: (gene. exp.) #2: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg#3: Chemical | 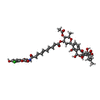  Mass: 932.275 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C46H42ClNO18 Mass: 932.275 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C46H42ClNO18#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 96 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 96 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.18 Å3/Da / Density % sol: 70.6 % / Description: NONE |
|---|---|
| Crystal grow | pH: 8.5 Details: 6% (W/V) PEG 8000, 30% (V/V) GLYCEROL IN 100 MM TRIS-HCL PH 8.5. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH3R / Wavelength: 1.542 |
| Detector | Type: MAR scanner 345 mm plate / Detector: IMAGE PLATE / Date: Nov 12, 2007 / Details: MIRRORS |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.542 Å / Relative weight: 1 |
| Reflection | Resolution: 2.62→27.47 Å / Num. obs: 58959 / % possible obs: 99.8 % / Observed criterion σ(I): -9 / Redundancy: 5.1 % / Biso Wilson estimate: 46.8 Å2 / Rmerge(I) obs: 0.09 / Net I/σ(I): 14 |
| Reflection shell | Resolution: 2.62→2.76 Å / Redundancy: 4.8 % / Rmerge(I) obs: 0.51 / Mean I/σ(I) obs: 3.3 / % possible all: 99.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1AB4 Resolution: 2.62→27.47 Å / Cor.coef. Fo:Fc: 0.942 / Cor.coef. Fo:Fc free: 0.917 / SU B: 21.13 / SU ML: 0.197 / Cross valid method: THROUGHOUT / ESU R: 0.309 / ESU R Free: 0.243 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. THE SIMOCYCLINONE D8 EFFECTIVELY CROSSLINKS A PAIR OF GYRA59 DIMERS ACROSS A CRYSTALLOGRAPHIC 2-FOLD AXIS. THE RESULTANT COMPLEX CONTAINS 4 ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. THE SIMOCYCLINONE D8 EFFECTIVELY CROSSLINKS A PAIR OF GYRA59 DIMERS ACROSS A CRYSTALLOGRAPHIC 2-FOLD AXIS. THE RESULTANT COMPLEX CONTAINS 4 MOLECULES OF SIMOCYCLINONE D8, WITH EACH LIGAND MAKING CONTACT WITH SUBUNITS FROM OPPOSING DIMERS SUCH THAT EACH GYRA59 SUBUNIT BINDS THE POLYKETIDE AND THE AMINOCOUMARIN MOIETIES, RESPECTIVELY, OF TWO SEPARATE SIMOCYCLINON D8 MOLECULES. THE TETRAMER IS ALSO OBSERVED IN SOLUTION AT HIGH LIGAND TO PROTEIN RATIOS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 52.1 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.62→27.47 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|