 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2ull | ||||||
|---|---|---|---|---|---|---|---|
| Title | MULTIPLE CONFORMATION STRUCTURE OF ALPHA-LYTIC PROTEASE AT 120 K | ||||||
 Components Components | ALPHA-LYTIC PROTEASE | ||||||
 Keywords Keywords | SERINE PROTEASE / HYDROLASE / ZYMOGEN / PROTEASE PRECURSOR | ||||||
| Function / homology |  Function and homology information Function and homology informationalpha-lytic endopeptidase / serine-type endopeptidase activity / proteolysis / extracellular region Similarity search - Function | ||||||
| Biological species |  Lysobacter enzymogenes (bacteria) Lysobacter enzymogenes (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / REFINEMENT OF 2ALP / Resolution: 1.5 Å X-RAY DIFFRACTION / SYNCHROTRON / REFINEMENT OF 2ALP / Resolution: 1.5 Å | ||||||
 Authors Authors | Rader, S.D. / Agard, D.A. | ||||||
 Citation Citation | Journal: Protein Sci. / Year: 1997 Title: Conformational substates in enzyme mechanism: the 120 K structure of alpha-lytic protease at 1.5 A resolution. Authors: Rader, S.D. / Agard, D.A. #1: Journal: Biochemistry / Year: 1991Title: Structural Basis for Broad Specificity in Alpha-Lytic Protease Mutants Authors: Bone, R. / Fujishige, A. / Kettner, C.A. / Agard, D.A. #2: Journal: Nature / Year: 1989Title: Structural Plasticity Broadens the Specificity of an Engineered Protease Authors: Bone, R. / Silen, J.L. / Agard, D.A. #3: Journal: Biochemistry / Year: 1989Title: Structural Analysis of Specificity: Alpha-Lytic Protease Complexes with Analogues of Reaction Intermediates Authors: Bone, R. / Frank, D. / Kettner, C.A. / Agard, D.A. #4: Journal: Biochemistry / Year: 1987Title: Serine Protease Mechanism: Structure of an Inhibitory Complex of Alpha-Lytic Protease and a Tightly Bound Peptide Boronic Acid Authors: Bone, R. / Shenvi, A.B. / Kettner, C.A. / Agard, D.A. #5: Journal: J.Mol.Biol. / Year: 1985Title: Refined Structure of Alpha-Lytic Protease at 1.7 A Resolution. Analysis of Hydrogen Bonding and Solvent Structure Authors: Fujinaga, M. / Delbaere, L.T. / Brayer, G.D. / James, M.N. #6: Journal: J.Mol.Biol. / Year: 1979Title: Molecular Structure of the Alpha-Lytic Protease from Myxobacter 495 at 2.8 Angstroms Resolution Authors: Brayer, G.D. / Delbaere, L.T. / James, M.N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2ull.cif.gz | 554.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2ull.ent.gz | 480.5 KB | Display | PDB format |
| PDBx/mmJSON format | 2ull.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ul/2ullftp://data.pdbj.org/pub/pdb/validation_reports/ul/2ull | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1talC  2alpS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |







-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Number of models | 16 |
-Components
| #1: Protein | Mass: 19875.131 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: THE ELECTRON DENSITY WAS MODELED WITH 16 CONFORMATIONS OF THE ENTIRE PROTEIN Source: (gene. exp.) Lysobacter enzymogenes (bacteria) / Production host: | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical |   Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4#3: Chemical | ChemComp-TAM / | 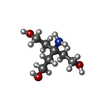  Mass: 163.215 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H17NO3 / Comment: pH buffer*YM Mass: 163.215 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H17NO3 / Comment: pH buffer*YM#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 301 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 301 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.5 Å3/Da / Density % sol: 48.2 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 8 / Details: pH 8.0 | ||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 7.5 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 120 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL7-1 / Wavelength: 1.08 / Beamline: BL7-1 / Wavelength: 1.08 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Aug 21, 1992 / Details: DUAL SLIT |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.08 Å / Relative weight: 1 |
| Reflection | Resolution: 1.5→20 Å / Num. obs: 33422 / % possible obs: 99 % / Observed criterion σ(I): -3 / Redundancy: 4.7 % / Biso Wilson estimate: 5.8 Å2 / Rsym value: 0.059 |
| Reflection shell | Resolution: 1.5→1.57 Å / % possible all: 99.1 |
| Reflection | *PLUS Num. measured all: 148867 / Rmerge(I) obs: 0.059 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: REFINEMENT OF 2ALP Starting model: PDB ENTRY 2ALP Resolution: 1.5→6 Å / Isotropic thermal model: FIXED B'S / Cross valid method: THROUGHOUT / σ(F): 0 Details: ELECTRON DENSITY WAS MODELED WITH 16 CONFORMATIONS OF THE ENTIRE PROTEIN. OCCUPANCIES FOR ALL ATOMS WERE SET TO 1/16. EACH CONFORMATION IS REPRESENTED AS A SEPARATE MODEL. THE DEPOSITOR ...Details: ELECTRON DENSITY WAS MODELED WITH 16 CONFORMATIONS OF THE ENTIRE PROTEIN. OCCUPANCIES FOR ALL ATOMS WERE SET TO 1/16. EACH CONFORMATION IS REPRESENTED AS A SEPARATE MODEL. THE DEPOSITOR PROVIDED ONE SET OF HETATM RECORDS. IN ORDER TO FOLLOW PDB FORMAT REQUIREMENTS, THE SAME HETATM RECORDS WERE INCLUDED IN EACH MODEL. NOTE THAT THE OCCUPANCY OF THE HETATM RECORDS WAS NOT DIVIDED BY 16.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 5.4 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.5→6 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.5→1.57 Å / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.155 / Rfactor Rfree: 0.189 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
