 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-24674 | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Seipin forms a flexible cage at lipid droplet formation sites | ||||||||||||||||||
 Map data Map data | |||||||||||||||||||
 Sample Sample |
| ||||||||||||||||||
 Keywords Keywords | lipid droplets / lipid droplet formation / complex / endoplasmic reticulum / fat storage / MEMBRANE PROTEIN | ||||||||||||||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation of sphingolipid biosynthetic process / lipid droplet formation / cortical endoplasmic reticulum / acylglycerol homeostasis / phospholipid homeostasis / lipid droplet organization / protein localization to lipid droplet / regulation of lipid metabolic process / positive regulation of lipid biosynthetic process / lipid droplet ...negative regulation of sphingolipid biosynthetic process / lipid droplet formation / cortical endoplasmic reticulum / acylglycerol homeostasis / phospholipid homeostasis / lipid droplet organization / protein localization to lipid droplet / regulation of lipid metabolic process / positive regulation of lipid biosynthetic process / lipid droplet / lipid metabolic process / intracellular protein localization / endoplasmic reticulum membrane / endoplasmic reticulum / identical protein binding Similarity search - Function | ||||||||||||||||||
| Biological species |  | ||||||||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.45 Å | ||||||||||||||||||
 Authors Authors | Arlt H / Sui X | ||||||||||||||||||
| Funding support |  United States, 5 items United States, 5 items
| ||||||||||||||||||
 Citation Citation | Journal: Nat Struct Mol Biol / Year: 2022 Title: Seipin forms a flexible cage at lipid droplet formation sites. Authors: Henning Arlt / Xuewu Sui / Brayden Folger / Carson Adams / Xiao Chen / Roman Remme / Fred A Hamprecht / Frank DiMaio / Maofu Liao / Joel M Goodman / Robert V Farese / Tobias C Walther /  Abstract: Lipid droplets (LDs) form in the endoplasmic reticulum by phase separation of neutral lipids. This process is facilitated by the seipin protein complex, which consists of a ring of seipin monomers, ...Lipid droplets (LDs) form in the endoplasmic reticulum by phase separation of neutral lipids. This process is facilitated by the seipin protein complex, which consists of a ring of seipin monomers, with a yet unclear function. Here, we report a structure of S. cerevisiae seipin based on cryogenic-electron microscopy and structural modeling data. Seipin forms a decameric, cage-like structure with the lumenal domains forming a stable ring at the cage floor and transmembrane segments forming the cage sides and top. The transmembrane segments interact with adjacent monomers in two distinct, alternating conformations. These conformations result from changes in switch regions, located between the lumenal domains and the transmembrane segments, that are required for seipin function. Our data indicate a model for LD formation in which a closed seipin cage enables triacylglycerol phase separation and subsequently switches to an open conformation to allow LD growth and budding. | ||||||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
 UCSF Chimera
UCSF Chimera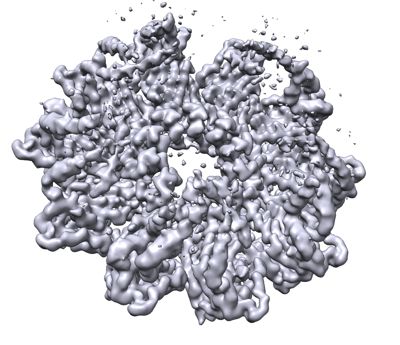
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_24674.map.gz | 77 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-24674-v30.xmlemd-24674.xml | 14.7 KB 14.7 KB | Display Display | EMDB header |
| Images |  emd_24674.png emd_24674.png | 158.6 KB | ||
| Filedesc metadata | emd-24674.cif.gz | 5.7 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-24674ftp://ftp.pdbj.org/pub/emdb/structures/EMD-24674 http://ftp.pdbj.org/pub/emdb/structures/EMD-24674ftp://ftp.pdbj.org/pub/emdb/structures/EMD-24674 | HTTPS FTP |
-Validation report
| Summary document | emd_24674_validation.pdf.gz | 522.5 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_24674_full_validation.pdf.gz | 522.1 KB | Display | |
| Data in XML | emd_24674_validation.xml.gz | 6.4 KB | Display | |
| Data in CIF | emd_24674_validation.cif.gz | 7.3 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-24674ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-24674 | HTTPS FTP |
-Related structure data
| Related structure data |  7rslMC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |





-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_24674.map.gz / Format: CCP4 / Size: 98.9 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.825 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
- Sample components
Sample components
-Entire : Seipin; Sei1; Fld1
| Entire | Name: Seipin; Sei1; Fld1 |
|---|---|
| Components |
|
-Supramolecule #1: Seipin; Sei1; Fld1
| Supramolecule | Name: Seipin; Sei1; Fld1 / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all Details: Seipin complex; 10 subunits of monomers (chain A-J) in two alternating conformations |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 66 KDa |
-Supramolecule #2: Seipin dimer
| Supramolecule | Name: Seipin dimer / type: complex / ID: 2 / Parent: 1 / Macromolecule list: all Details: Dimer of seipin monomers in transmembrane segment conformation A and B (chain A and B). That was used to build the oligomer consisting of 5 dimers (10 monomers). |
|---|---|
| Source (natural) | Organism: |
-Macromolecule #1: Seipin
| Macromolecule | Name: Seipin / type: protein_or_peptide / ID: 1 / Number of copies: 10 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 32.623953 KDa |
| Recombinant expression | Organism: |
| Sequence | String: MKINVSRPLQ FLQWSSYIVV AFLIQLLIIL PLSILIYHDF YLRLLPADSS NVVPLNTFNI LNGVQFGTKF FQSIKSIPVG TDLPQTIDN GLSQLIPMRD NMEYKLDLNL QLYCQSKTDH LNLDNLLIDV YRGPGPLLGA PGGSNSKDEK IFHTSRPIVC L ALTDSMSP ...String: MKINVSRPLQ FLQWSSYIVV AFLIQLLIIL PLSILIYHDF YLRLLPADSS NVVPLNTFNI LNGVQFGTKF FQSIKSIPVG TDLPQTIDN GLSQLIPMRD NMEYKLDLNL QLYCQSKTDH LNLDNLLIDV YRGPGPLLGA PGGSNSKDEK IFHTSRPIVC L ALTDSMSP QEIEQLGPSR LDVYDEEWLN TIRIEDKISL ESSYETISVF LKTEIAQRNL IIHPESGIKF RMNFEQGLRN LM LRKRFLS YIIGISIFHC IICVLFFITG CTAFIFVRKG QEKSKKHS UniProtKB: Seipin |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 3.2 mg/mL | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 8 Component:
| ||||||||||||
| Grid | Model: Quantifoil R1.2/1.3 / Material: COPPER / Mesh: 400 / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 30 sec. / Pretreatment - Atmosphere: AIR | ||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV | ||||||||||||
| Details | This sample was monodisperse. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K3 (6k x 4k) / Number grids imaged: 1 / Number real images: 12655 / Average exposure time: 1.9 sec. / Average electron dose: 54.59 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source: OTHER |
| Electron optics | C2 aperture diameter: 50.0 µm / Illumination mode: OTHER / Imaging mode: OTHER / Nominal defocus max: 2.5 µm / Nominal defocus min: 1.2 µm / Nominal magnification: 105000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing
-Atomic model buiding 1
| Refinement | Protocol: AB INITIO MODEL |
|---|---|
| Output model | PDB-7rsl: |
