 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1bda: CATALYTIC DOMAIN OF HUMAN SINGLE CHAIN TISSUE PLASMINOGEN ACTIVAT... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1bda | ||||||
|---|---|---|---|---|---|---|---|
| Title | CATALYTIC DOMAIN OF HUMAN SINGLE CHAIN TISSUE PLASMINOGEN ACTIVATOR IN COMPLEX WITH DANSYL-EGR-CMK (DANSYL-GLU-GLY-ARG CHLOROMETHYL KETONE) | ||||||
 Components Components | SINGLE CHAIN TISSUE TYPE PLASMINOGEN ACTIVATOR | ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / TRYPSIN LIKE SERINE PROTEASE / FIBRINOLYTIC ENZYMES / PLASMINOGEN ACTIVATORS / HYDROLASE-HYDROLASE INHIBITOR COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationt-plasminogen activator / prevention of polyspermy / trans-synaptic signaling by BDNF, modulating synaptic transmission / Signaling by PDGF / negative regulation of plasminogen activation / Dissolution of Fibrin Clot / smooth muscle cell migration / plasminogen activation / platelet-derived growth factor receptor signaling pathway / negative regulation of fibrinolysis ...t-plasminogen activator / prevention of polyspermy / trans-synaptic signaling by BDNF, modulating synaptic transmission / Signaling by PDGF / negative regulation of plasminogen activation / Dissolution of Fibrin Clot / smooth muscle cell migration / plasminogen activation / platelet-derived growth factor receptor signaling pathway / negative regulation of fibrinolysis / serine protease inhibitor complex / fibrinolysis / negative regulation of proteolysis / secretory granule / protein modification process / phosphoprotein binding / Schaffer collateral - CA1 synapse / apical part of cell / blood coagulation / response to hypoxia / serine-type endopeptidase activity / signaling receptor binding / glutamatergic synapse / cell surface / proteolysis / : / extracellular exosome / extracellular region / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 3.35 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 3.35 Å | ||||||
 Authors Authors | Bode, W. / Renatus, M. / Engh, R.A. | ||||||
 Citation Citation | Journal: EMBO J. / Year: 1997 Title: Lysine 156 promotes the anomalous proenzyme activity of tPA: X-ray crystal structure of single-chain human tPA. Authors: Renatus, M. / Engh, R.A. / Stubbs, M.T. / Huber, R. / Fischer, S. / Kohnert, U. / Bode, W. #1: Journal: Biochemistry / Year: 1997Title: Catalytic Domain Structure of Vampire Bat Plasminogen Activator: A Molecular Paradigm for Proteolysis without Activation Cleavage Authors: Renatus, M. / Stubbs, M.T. / Huber, R. / Bringmann, P. / Donner, P. / Schleuning, W.D. / Bode, W. #2: Journal: J.Mol.Biol. / Year: 1996Title: The 2.3 A Crystal Structure of the Catalytic Domain of Recombinant Two-Chain Human Tissue-Type Plasminogen Activator Authors: Lamba, D. / Bauer, M. / Huber, R. / Fischer, S. / Rudolph, R. / Kohnert, U. / Bode, W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1bda.cif.gz | 119.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1bda.ent.gz | 92.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1bda.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bd/1bdaftp://data.pdbj.org/pub/pdb/validation_reports/bd/1bda | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1rtfS S: Starting model for refinement |
|---|---|
| Similar structure data |









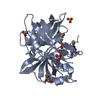
-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||
| 2 | 
| ||||||||||||
| Unit cell |
| ||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
| #1: Protein | Mass: 29725.654 Da / Num. of mol.: 2 / Fragment: UNP residues 298-562 / Mutation: DELETION MUTANT Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Production host:  #2: Chemical | 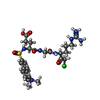  Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 627.133 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C26H37ClN7O7S Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 627.133 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C26H37ClN7O7SReferences: dansylglutamyl-glycyl-arginine chloromethyl ketone #3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 19 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 19 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | Nonpolymer details | THE UNBOUND FORM OF THE INHIBITOR IS DANSYL-GLU-GLY-ARG-CMK-CHLOROMETHYLKETONE. UPON REACTION WITH ...THE UNBOUND FORM OF THE INHIBITOR IS DANSYL-GLU-GLY-ARG-CMK-CHLOROMETH | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.36 Å3/Da / Density % sol: 48 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 8.2 / Details: pH 8.2 | ||||||||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS Density % sol: 50 % | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 23 ℃ / Method: vapor diffusion, sitting drop | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 290 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH2R / Wavelength: 1.5418 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jun 13, 1996 |
| Radiation | Monochromator: GRAPHITE(002) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 3.1→25 Å / Num. obs: 9414 / % possible obs: 86.7 % / Observed criterion σ(I): 2 / Redundancy: 2.9 % / Rmerge(I) obs: 0.147 / Net I/σ(I): 3.6 |
| Reflection shell | Resolution: 3.1→3.18 Å / Redundancy: 1.7 % / Rmerge(I) obs: 0.63 / Mean I/σ(I) obs: 1.2 / % possible all: 55.2 |
| Reflection | *PLUS Num. measured all: 27265 |
| Reflection shell | *PLUS % possible obs: 55.2 % / Rmerge(I) obs: 0.63 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1RTF Resolution: 3.35→7 Å / Data cutoff high absF: 100000 / Data cutoff low absF: 0.25 / Isotropic thermal model: RESTRAINED / Cross valid method: NO FREE R-FACTOR / σ(F): 2 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 26.1 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.35→7 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 3.25→3.35 Å / Total num. of bins used: 10
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.851 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.2037 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
