| 登録情報 | データベース: PDB / ID: 1xvg
|
|---|
| タイトル | soluble methane monooxygenase hydroxylase: bromoethanol soaked structure |
|---|
 要素 要素 | (Methane monooxygenase component A ...) x 3 |
|---|
 キーワード キーワード | OXIDOREDUCTASE / methane / diiron / product binding / cavities / four-helix bundle |
|---|
| 機能・相同性 |  機能・相同性情報 機能・相同性情報
methane metabolic process / methane monooxygenase (soluble) / methane monooxygenase [NAD(P)H] activity / one-carbon metabolic process / metal ion binding類似検索 - 分子機能 Methane monooxygenase, gamma chain, domain 1 / Methane monooxygenase, gamma chain, domain 2 / Methane monooxygenase, gamma chain / Methane monooxygenase, gamma chain, domain 1 / Methane monooxygenase, gamma chain, domain 2 / Methane monooxygenase, gamma chain superfamily / : / : / Methane monooxygenase, hydrolase gamma chain / Methane/phenol monooxygenase, hydroxylase component ...Methane monooxygenase, gamma chain, domain 1 / Methane monooxygenase, gamma chain, domain 2 / Methane monooxygenase, gamma chain / Methane monooxygenase, gamma chain, domain 1 / Methane monooxygenase, gamma chain, domain 2 / Methane monooxygenase, gamma chain superfamily / : / : / Methane monooxygenase, hydrolase gamma chain / Methane/phenol monooxygenase, hydroxylase component / Propane/methane/phenol/toluene hydroxylase / Methane/Phenol/Alkene Hydroxylase / Ribonucleotide Reductase, subunit A / Ribonucleotide Reductase, subunit A / Monooxygenase / Ribonucleotide reductase-like / Ferritin-like superfamily / Up-down Bundle / Orthogonal Bundle / Mainly Alpha類似検索 - ドメイン・相同性 2-BROMOETHANOL / : / Methane monooxygenase component A gamma chain / Methane monooxygenase component A beta chain / Methane monooxygenase component A alpha chain類似検索 - 構成要素 |
|---|
| 生物種 |  Methylococcus capsulatus (バクテリア) Methylococcus capsulatus (バクテリア) |
|---|
| 手法 |  X線回折 / シンクロトロン / 分子置換 / 解像度: 1.96 Å X線回折 / シンクロトロン / 分子置換 / 解像度: 1.96 Å |
|---|
 データ登録者 データ登録者 | Sazinsky, M.H. / Lippard, S.J. |
|---|
 引用 引用 | ジャーナル: J.Am.Chem.Soc. / 年: 2005
タイトル: Product Bound Structures of the Soluble Methane Monooxygenase Hydroxylase from Methylococcus capsulatus (Bath): Protein Motion in the Alpha-Subunit
著者: Sazinsky, M.H. / Lippard, S.J. |
|---|
| 履歴 | | 登録 | 2004年10月27日 | 登録サイト: RCSB / 処理サイト: RCSB |
|---|
| 改定 1.0 | 2005年5月3日 | Provider: repository / タイプ: Initial release |
|---|
| 改定 1.1 | 2007年10月16日 | Group: Version format compliance |
|---|
| 改定 1.2 | 2011年7月13日 | Group: Derived calculations / Version format compliance |
|---|
| 改定 1.3 | 2023年8月23日 | Group: Data collection / Database references ...Data collection / Database references / Derived calculations / Refinement description
カテゴリ: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model / pdbx_struct_conn_angle / struct_conn / struct_site
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.value / _struct_conn.pdbx_dist_value / _struct_conn.ptnr1_auth_asym_id / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_asym_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id |
|---|
|
|---|
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード

















 PDBj
PDBj
 集合体
集合体



 分子量: 55.845 Da / 分子数: 4 / 由来タイプ: 合成 / 式: Fe
分子量: 55.845 Da / 分子数: 4 / 由来タイプ: 合成 / 式: Fe 分子量: 40.078 Da / 分子数: 4 / 由来タイプ: 合成 / 式: Ca
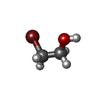
分子量: 40.078 Da / 分子数: 4 / 由来タイプ: 合成 / 式: Ca 分子量: 124.965 Da / 分子数: 12 / 由来タイプ: 合成 / 式: C2H5BrO
分子量: 124.965 Da / 分子数: 12 / 由来タイプ: 合成 / 式: C2H5BrO 試料調製
試料調製 / ビームライン: BL7-1 / 波長: 1.08 Å
/ ビームライン: BL7-1 / 波長: 1.08 Å 解析
解析