 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1pjp: THE 2.2 A CRYSTAL STRUCTURE OF HUMAN CHYMASE IN COMPLEX WITH SUCC... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1pjp | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | THE 2.2 A CRYSTAL STRUCTURE OF HUMAN CHYMASE IN COMPLEX WITH SUCCINYL-ALA-ALA-PRO-PHE-CHLOROMETHYLKETONE | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / HUMAN CHYMASE / SERINE PROTEINASE / DIPEPTIDYL CARBOXYPEPTIDASE / ANGIOTENSIN / HYDROLASE-HYDROLASE INHIBITOR COMPLEX | |||||||||
| Function / homology |  Function and homology information Function and homology informationchymase / cytokine precursor processing / basement membrane disassembly / peptide metabolic process / midbrain development / Activation of Matrix Metalloproteinases / extracellular matrix disassembly / angiotensin maturation / Metabolism of Angiotensinogen to Angiotensins / serine-type peptidase activity ...chymase / cytokine precursor processing / basement membrane disassembly / peptide metabolic process / midbrain development / Activation of Matrix Metalloproteinases / extracellular matrix disassembly / angiotensin maturation / Metabolism of Angiotensinogen to Angiotensins / serine-type peptidase activity / secretory granule / cellular response to glucose stimulus / peptide binding / protein catabolic process / Signaling by SCF-KIT / cytoplasmic ribonucleoprotein granule / positive regulation of angiogenesis / regulation of inflammatory response / collagen-containing extracellular matrix / endopeptidase activity / intracellular membrane-bounded organelle / serine-type endopeptidase activity / extracellular space / extracellular region / cytosol / cytoplasm Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human)synthetic construct (others) | |||||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.2 Å | |||||||||
 Authors Authors | Pereira, P.J.B. / Wang, Z.M. / Rubin, H. / Huber, R. / Bode, W. / Schechter, N.M. / Strobl, S. | |||||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 1999 Title: The 2.2 A crystal structure of human chymase in complex with succinyl-Ala-Ala-Pro-Phe-chloromethylketone: structural explanation for its dipeptidyl carboxypeptidase specificity. Authors: Pereira, P.J. / Wang, Z.M. / Rubin, H. / Huber, R. / Bode, W. / Schechter, N.M. / Strobl, S. #1: Journal: Biochemistry / Year: 1997Title: Crystal Structure of Phenylmethanesulfonyl Fluoride-Treated Human Chymase at 1.9 A Authors: McGrath, M.E. / Mirzadegan, T. / Schmidt, B.F. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1pjp.cif.gz | 65.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1pjp.ent.gz | 46.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1pjp.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1pjp_validation.pdf.gz | 399.2 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1pjp_full_validation.pdf.gz | 400.5 KB | Display | |
| Data in XML | 1pjp_validation.xml.gz | 6.5 KB | Display | |
| Data in CIF | 1pjp_validation.cif.gz | 10.1 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/pj/1pjpftp://data.pdbj.org/pub/pdb/validation_reports/pj/1pjp | HTTPS FTP |
-Related structure data
| Related structure data |  3rp2S S: Starting model for refinement |
|---|---|
| Similar structure data |




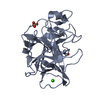





-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 24991.857 Da / Num. of mol.: 1 / Mutation: F127K, V208A, R235Q Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Cell: MAST CELLS / Gene: CMA1, CYH, CYM / Production host:   Spodoptera frugiperda (fall armyworm) / References: UniProt: P23946, chymase Spodoptera frugiperda (fall armyworm) / References: UniProt: P23946, chymase | ||||||
|---|---|---|---|---|---|---|---|
| #2: Protein/peptide | 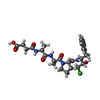  Type: Peptide-like / Class: Inhibitor / Mass: 539.021 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) / References: SUCCINYL-ALA-ALA-PRO-PHE-CHLOROMETHYLKETONE Type: Peptide-like / Class: Inhibitor / Mass: 539.021 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) / References: SUCCINYL-ALA-ALA-PRO-PHE-CHLOROMETHYLKETONE | ||||||
| #3: Sugar |   Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H15NO6 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H15NO6#4: Chemical | ChemComp-ZN / |   Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 144 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 144 / Source method: isolated from a natural source / Formula: H2OCompound details | THE UNBOUND FORM OF THE INHIBITOR IS SUCCINYL-ALA-ALA-PRO-PHE-CHLOROMETHYLKETONE. UPON REACTION ...THE UNBOUND FORM OF THE INHIBITOR IS SUCCINYL-ALA-ALA-PRO-PHE-CHLOROMETH | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.68 Å3/Da / Density % sol: 54 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7.5 / Details: pH 7.5 | ||||||||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 22 ℃ / Method: vapor diffusion, hanging drop / pH: 6.8 | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 301 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE |
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.2→20 Å / Num. obs: 14122 / % possible obs: 96.4 % / Redundancy: 9.78 % / Rmerge(I) obs: 0.076 |
| Reflection shell | Resolution: 2.2→2.26 Å / Rmerge(I) obs: 0.254 / % possible all: 98.8 |
| Reflection | *PLUS Num. measured all: 138168 |
| Reflection shell | *PLUS % possible obs: 98.8 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 3RP2 Resolution: 2.2→8 Å / Cross valid method: THROUGHOUT / σ(F): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 22.6 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.851 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.2 Å / Lowest resolution: 8 Å / σ(F): 2 / Num. reflection Rfree: 649 / % reflection Rfree: 5 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 22.6 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
