 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1mui | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of HIV-1 protease complexed with Lopinavir. | ||||||
 Components Components | protease | ||||||
 Keywords Keywords | HYDROLASE | ||||||
| Function / homology |  Function and homology information Function and homology informationhost multivesicular body / aspartic-type endopeptidase activity / virion membrane / proteolysis Similarity search - Function | ||||||
| Biological species |   Human immunodeficiency virus 1 Human immunodeficiency virus 1 | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 2.8 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 2.8 Å | ||||||
 Authors Authors | Stoll, V. / Qin, W. / Stewart, K.D. / Jakob, C. / Park, C. / Walter, K. / Simmer, R.L. / Helfrich, R. / Bussiere, D. / Kao, J. ...Stoll, V. / Qin, W. / Stewart, K.D. / Jakob, C. / Park, C. / Walter, K. / Simmer, R.L. / Helfrich, R. / Bussiere, D. / Kao, J. / Kempf, D. / Sham, H.L. / Norbeck, D.W. | ||||||
 Citation Citation | Journal: BIOORG.MED.CHEM. / Year: 2002 Title: X-ray Crystallographic Structure of ABT-378 (Lopinavir) Bound to HIV-1 Protease Authors: Stoll, V. / Qin, W. / Stewart, K.D. / Jakob, C. / Park, C. / Walter, K. / Simmer, R.L. / Helfrich, R. / Bussiere, D. / Kao, J. / Kempf, D. / Sham, H.L. / Norbeck, D.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1mui.cif.gz | 50.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1mui.ent.gz | 37.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1mui.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/mu/1muiftp://data.pdbj.org/pub/pdb/validation_reports/mu/1mui | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  9hvpS S: Starting model for refinement |
|---|---|
| Similar structure data |


















-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 10803.756 Da / Num. of mol.: 2 / Fragment: residues 57-155 / Mutation: N37S Source method: isolated from a genetically manipulated source Source: (gene. exp.) Human immunodeficiency virus 1 / Genus: Lentivirus / Plasmid: pET11b / Production host:  #2: Chemical | ChemComp-AB1 / | 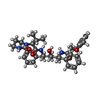  Mass: 628.801 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C37H48N4O5 Mass: 628.801 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C37H48N4O5 |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.23 Å3/Da / Density % sol: 44.95 % |
|---|---|
| Crystal grow | Temperature: 295 K / pH: 4.6 Details: 100mM Na acetate, 0.4-0.8M NaCl, pH 4.6, temperature 295K |
| Crystal grow | *PLUS Method: unknownDetails: Fizgerald, P.M.D., (1990) J. Biol. Chem., 265, 14209. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 17-ID / Beamline: 17-ID |
| Detector | Type: MARRESEARCH / Detector: CCD |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Relative weight: 1 |
| Reflection | Resolution: 2.8→33.24 Å / Num. obs: 4652 / % possible obs: 93.2 % / Observed criterion σ(F): 2 / Observed criterion σ(I): 2 / Biso Wilson estimate: -2.1 Å2 / Rsym value: 0.096 / Net I/σ(I): 9.2 |
| Reflection shell | Resolution: 2.8→2.9 Å / Rsym value: 0.345 / % possible all: 97.5 |
- Processing
Processing
| Software |
| |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: pdb code 9HVP Resolution: 2.8→33 Å / Isotropic thermal model: anisotropic / Cross valid method: THROUGHOUT / σ(F): 2 / Stereochemistry target values: Engh & Huber
| |||||||||||||||||||||
| Displacement parameters |
| |||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.5 Å / Luzzati sigma a obs: 0.43 Å | |||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→33 Å
| |||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||
| LS refinement shell | Resolution: 2.8→2.98 Å / Rfactor Rfree error: 0.048
| |||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 33 Å / % reflection Rfree: 7 % / Rfactor obs: 0.309 / Rfactor Rfree: 0.32 / Rfactor Rwork: 0.26 | |||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||
| Refine LS restraints | *PLUS
| |||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 2.8 Å / Rfactor Rfree: 0.322 / Rfactor Rwork: 0.276 |
