 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1mlz: Crystal Structure of 7,8-Diaminopelargonic Acid Synthase in compl... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1mlz | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of 7,8-Diaminopelargonic Acid Synthase in complex with the trans-isomer of amiclenomycin. | ||||||
 Components Components | 7,8-diamino-pelargonic acid aminotransferase | ||||||
 Keywords Keywords | TRANSFERASE / Aminotransferase / Fold type I / subclass II / amiclenomycin | ||||||
| Function / homology |  Function and homology information Function and homology informationadenosylmethionine-8-amino-7-oxononanoate transaminase / S-adenosyl-L-methionine:8-amino-7-oxononanoate transaminase activity / biotin biosynthetic process / pyridoxal phosphate binding / protein homodimerization activity / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.15 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.15 Å | ||||||
 Authors Authors | Sandmark, J. / Mann, S. / Marquet, A. / Schneider, G. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2002 Title: Structural basis for the inhibition of the biosynthesis of biotin by the antibiotic amiclenomycin Authors: Sandmark, J. / Mann, S. / Marquet, A. / Schneider, G. | ||||||
| History |
| ||||||
| Remark 999 | sequence The crystallized protein differs from the Swissprot sequence at residue 14. Number 14 is a ...sequence The crystallized protein differs from the Swissprot sequence at residue 14. Number 14 is a leucin, while in the Swissprot sequence number 14 is a tryptophan. This is confirmed by DNA sequencing and was also reported for the original WT structure (1qj5). |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1mlz.cif.gz | 185.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1mlz.ent.gz | 147.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1mlz.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ml/1mlzftp://data.pdbj.org/pub/pdb/validation_reports/ml/1mlz | HTTPS FTP |
|---|
-Related structure data















-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 47310.391 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) References: UniProt: P12995, adenosylmethionine-8-amino-7-oxononanoate transaminase #2: Chemical | 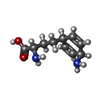  Mass: 196.246 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16N2O2 Mass: 196.246 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16N2O2#3: Chemical |   Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na#4: Chemical |   Mass: 247.142 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H10NO6P Mass: 247.142 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H10NO6P#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 324 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 324 / Source method: isolated from a natural source / Formula: H2ONonpolymer details | A covalent adduct is formed between pyridoxal-5'-phosphate and the amiclenomycin in the A subunit. ...A covalent adduct is formed between pyridoxal-5'-phosphate and the amiclenomycin in the A subunit. In the B subunit the density for amiclenomycin is significantly weaker. The amiclenomycin molecule is modeled as non-covalently bound in the B subunit, but the model has high B factors and is not well supported by structural data. For a better view of the pyridoxal-5'-phosphate-amiclenomycin adduct see the A subunit in the structure of the enzyme in complex with the cis-isomer (PDB ID 1mly). For details see the primary citation. | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.07 Å3/Da / Density % sol: 40.49 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 294 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: PEG4000, MPD, HEPES, pH 7.5, VAPOR DIFFUSION, HANGING DROP, temperature 294K | ||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion / Details: Kack, H., (1998) Acta Crystallogr., D54, 1397. | ||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: MAX II  / Beamline: I711 / Wavelength: 0.991 Å / Beamline: I711 / Wavelength: 0.991 Å |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: May 10, 2001 |
| Radiation | Monochromator: Si(111) monochromator crystal / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.991 Å / Relative weight: 1 |
| Reflection | Resolution: 2.15→22.36 Å / Num. all: 41748 / Num. obs: 41748 / % possible obs: 98.6 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Biso Wilson estimate: 31.2 Å2 / Rsym value: 0.066 / Net I/σ(I): 13.9 |
| Reflection shell | Resolution: 2.15→2.27 Å / Mean I/σ(I) obs: 2.9 / Rsym value: 0.291 / % possible all: 96 |
| Reflection | *PLUS Lowest resolution: 20 Å / Num. obs: 38070 / Num. measured all: 112317 / Rmerge(I) obs: 0.066 |
| Reflection shell | *PLUS % possible obs: 96 % / Rmerge(I) obs: 0.291 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.15→22.36 Å / Cor.coef. Fo:Fc: 0.945 / Cor.coef. Fo:Fc free: 0.923 / SU B: 9.138 / SU ML: 0.237 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.334 / ESU R Free: 0.219 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: In monomer A two stretches of residues are disordered, 159-169 and 186-192 resp. In monomer B three stretches of residues are disordered, 158-168, 189-194 and 296-301 resp. Residue 429 was ...Details: In monomer A two stretches of residues are disordered, 159-169 and 186-192 resp. In monomer B three stretches of residues are disordered, 158-168, 189-194 and 296-301 resp. Residue 429 was excluded from both monomers. A number of sidechains on the surface of the protein are disordered. The occupancy for these is estimated to 0 in most cases.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 32.347 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.15→22.36 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.15→2.205 Å / Total num. of bins used: 20 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 20 Å / Rfactor Rfree: 0.24 / Rfactor Rwork: 0.205 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
