| ソフトウェア | | 名称 | バージョン | 分類 |
|---|
| REFMAC | 5.1.05| 精密化 | | dtDisplay | | データ収集 | | HKL-2000 | | データ削減 | | CNS | | 精密化 | | d*TREK | | データスケーリング | | d*TREK | | データ削減 | | DTDISPLAY | | データ削減 | | HKL-2000 | | データスケーリング | | CNS | | 位相決定 | |
|
|---|
| 精密化 | 構造決定の手法:  多波長異常分散 / 解像度: 1.7→74.54 Å / Cor.coef. Fo:Fc: 0.972 / Cor.coef. Fo:Fc free: 0.952 / SU B: 1.933 / SU ML: 0.062 / TLS residual ADP flag: LIKELY RESIDUAL / 交差検証法: THROUGHOUT / σ(F): 0 / ESU R: 0.099 / ESU R Free: 0.102 / 立体化学のターゲット値: MAXIMUM LIKELIHOOD / 詳細: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS 多波長異常分散 / 解像度: 1.7→74.54 Å / Cor.coef. Fo:Fc: 0.972 / Cor.coef. Fo:Fc free: 0.952 / SU B: 1.933 / SU ML: 0.062 / TLS residual ADP flag: LIKELY RESIDUAL / 交差検証法: THROUGHOUT / σ(F): 0 / ESU R: 0.099 / ESU R Free: 0.102 / 立体化学のターゲット値: MAXIMUM LIKELIHOOD / 詳細: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| Rfactor | 反射数 | %反射 | Selection details |
|---|
| Rfree | 0.18838 | 1422 | 5 % | RANDOM |
|---|
| Rwork | 0.14523 | - | - | - |
|---|
| obs | 0.14735 | 27141 | 93.85 % | - |
|---|
| all | - | 30435 | - | - |
|---|
|
|---|
| 溶媒の処理 | イオンプローブ半径: 0.8 Å / 減衰半径: 0.8 Å / VDWプローブ半径: 1.4 Å / 溶媒モデル: BABINET MODEL WITH MASK |
|---|
| 原子変位パラメータ | Biso mean: 15.249 Å2
| Baniso -1 | Baniso -2 | Baniso -3 |
|---|
| 1- | -0.15 Å2 | 0 Å2 | 0 Å2 |
|---|
| 2- | - | -0.15 Å2 | 0 Å2 |
|---|
| 3- | - | - | 0.3 Å2 |
|---|
|
|---|
| 精密化ステップ | サイクル: LAST / 解像度: 1.7→74.54 Å
| タンパク質 | 核酸 | リガンド | 溶媒 | 全体 |
|---|
| 原子数 | 1996 | 0 | 14 | 238 | 2248 |
|---|
|
|---|
| 拘束条件 | | Refine-ID | タイプ | Dev ideal | Dev ideal target | 数 |
|---|
| X-RAY DIFFRACTION | r_bond_refined_d| 0.022 | 0.021 | 2327 | | X-RAY DIFFRACTION | r_bond_other_d| 0.003 | 0.02 | 2174 | | X-RAY DIFFRACTION | r_angle_refined_deg| 1.941 | 1.95 | 3158 | | X-RAY DIFFRACTION | r_angle_other_deg| 2.329 | 3 | 5079 | | X-RAY DIFFRACTION | r_dihedral_angle_1_deg| 6.629 | 5 | 276 | | X-RAY DIFFRACTION | r_dihedral_angle_3_deg| 19.565 | 15 | 455 | | X-RAY DIFFRACTION | r_chiral_restr| 0.143 | 0.2 | 352 | | X-RAY DIFFRACTION | r_gen_planes_refined| 0.011 | 0.02 | 2551 | | X-RAY DIFFRACTION | r_gen_planes_other| 0.009 | 0.02 | 468 | | X-RAY DIFFRACTION | r_nbd_refined| 0.286 | 0.2 | 539 | | X-RAY DIFFRACTION | r_nbd_other| 0.268 | 0.2 | 2506 | | X-RAY DIFFRACTION | r_nbtor_other| 0.098 | 0.2 | 1273 | | X-RAY DIFFRACTION | r_xyhbond_nbd_refined| 0.196 | 0.2 | 144 | | X-RAY DIFFRACTION | r_symmetry_vdw_refined| 0.3 | 0.2 | 17 | | X-RAY DIFFRACTION | r_symmetry_vdw_other| 0.293 | 0.2 | 59 | | X-RAY DIFFRACTION | r_symmetry_hbond_refined| 0.142 | 0.2 | 18 | | X-RAY DIFFRACTION | r_mcbond_it| 1.107 | 1.5 | 1373 | | X-RAY DIFFRACTION | r_mcangle_it| 1.791 | 2 | 2248 | | X-RAY DIFFRACTION | r_scbond_it| 2.65 | 3 | 954 | | X-RAY DIFFRACTION | r_scangle_it| 3.961 | 4.5 | 910 | | | | | | | | | | | | | | | | | | | | |
|
|---|
| LS精密化 シェル | 解像度: 1.7→1.792 Å / Total num. of bins used: 10 / | Rfactor | 反射数 |
|---|
| Rfree | 0.256 | 175 |
|---|
| Rwork | 0.212 | 3631 |
|---|
|
|---|
| 精密化 TLS | 手法: refined / Refine-ID: X-RAY DIFFRACTION | ID | L11 (°2) | L12 (°2) | L13 (°2) | L22 (°2) | L23 (°2) | L33 (°2) | S11 (Å °) | S12 (Å °) | S13 (Å °) | S21 (Å °) | S22 (Å °) | S23 (Å °) | S31 (Å °) | S32 (Å °) | S33 (Å °) | T11 (Å2) | T12 (Å2) | T13 (Å2) | T22 (Å2) | T23 (Å2) | T33 (Å2) | Origin x (Å) | Origin y (Å) | Origin z (Å) |
|---|
| 1 | 1.596 | -0.2217 | 0.2992 | 2.1184 | -0.0851 | 1.7319 | 0.0179 | 0.0109 | 0.0518 | 0.0451 | -0.0312 | 0.0583 | -0.0046 | -0.026 | 0.0133 | 0.021 | 0.0147 | 0.0092 | 0.019 | -0.0111 | 0.0393 | 18.9707 | 10.823 | 14.3217 | | 2 | 0.9386 | -0.4449 | -0.1438 | 1.3232 | -0.4501 | 1.1053 | 0.0483 | 0.2257 | -0.0386 | -0.1064 | -0.0571 | -0.3146 | 0.0521 | 0.3817 | 0.0088 | 0.1338 | 0.0224 | -0.0063 | 0.162 | -0.0055 | 0.1404 | 34.4722 | 2.5709 | 11.6658 | | 3 | 1.6269 | -0.3573 | 0.0707 | 1.8027 | -0.1525 | 1.4051 | 0.0233 | 0.0193 | 0.0915 | 0.0099 | -0.0147 | -0.0578 | -0.0585 | 0.0426 | -0.0086 | 0.171 | 0.0069 | -0.0114 | 0.1592 | -0.0155 | 0.1408 | 22.4028 | 9.3391 | 13.223 |
|
|---|
| 精密化 TLSグループ | | ID | Refine-ID | Refine TLS-ID | Auth asym-ID | Label asym-ID | Auth seq-ID | Label seq-ID |
|---|
| 1 | X-RAY DIFFRACTION | 1 | AA| 3 - 109 | 3 - 109 | | 2 | X-RAY DIFFRACTION | 1 | AA| 188 - 258 | 188 - 258 | | 3 | X-RAY DIFFRACTION | 2 | AA| 110 - 187 | 110 - 187 | | 4 | X-RAY DIFFRACTION | 3 | AA| 303 - 540 | 1 - 238 | | | | | | | | |
|
|---|
| 精密化 | *PLUS 最低解像度: 75 Å / Rfactor Rfree: 0.189 / Rfactor Rwork: 0.147 |
|---|
| 溶媒の処理 | *PLUS |
|---|
| 原子変位パラメータ | *PLUS |
|---|
| 拘束条件 | *PLUS | Refine-ID | タイプ | Dev ideal | Dev ideal target |
|---|
| X-RAY DIFFRACTION | r_bond_d| 0.022 | 0.021 | | X-RAY DIFFRACTION | r_angle_d | | | X-RAY DIFFRACTION | r_angle_deg| 1.905 | 1.95 | | X-RAY DIFFRACTION | r_plane_restr| 0.011 | 0.02 | | X-RAY DIFFRACTION | r_chiral_restr| 0.131 | 0.2 | | | | | |
|
|---|
| LS精密化 シェル | *PLUS 最低解像度: 1.79 Å / Rfactor Rfree: 0.246 |
|---|
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報
 データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード









 PDBj
PDBj


 集合体
集合体
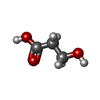
 分子量: 90.078 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C3H6O3
分子量: 90.078 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C3H6O3
 分子量: 62.068 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C2H6O2
分子量: 62.068 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C2H6O2 分子量: 18.015 Da / 分子数: 238 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 238 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: 19-ID / 波長: 0.979464, 0.953732, 1.03321
/ ビームライン: 19-ID / 波長: 0.979464, 0.953732, 1.03321 解析
解析