 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 1igb | ||||||
|---|---|---|---|---|---|---|---|
| タイトル | AEROMONAS PROTEOLYTICA AMINOPEPTIDASE COMPLEXED WITH THE INHIBITOR PARA-IODO-D-PHENYLALANINE HYDROXAMATE | ||||||
 要素 要素 | AMINOPEPTIDASE | ||||||
 キーワード キーワード | AMINOPEPTIDASE / HYDROLASE | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報bacterial leucyl aminopeptidase / metalloexopeptidase activity / aminopeptidase activity / proteolysis / extracellular region / metal ion binding 類似検索 - 分子機能 | ||||||
| 生物種 |  Vibrio proteolyticus (バクテリア) Vibrio proteolyticus (バクテリア) | ||||||
| 手法 |  X線回折 / MOLECULAR REPLACEMENT SOFTWARE USED : X-PLOR STARTING MODEL FOR MOLECULAR REPLACEMENT: NATIVE PROTEIN (REFERENCE 1) / 解像度: 2.3 Å X線回折 / MOLECULAR REPLACEMENT SOFTWARE USED : X-PLOR STARTING MODEL FOR MOLECULAR REPLACEMENT: NATIVE PROTEIN (REFERENCE 1) / 解像度: 2.3 Å | ||||||
 データ登録者 データ登録者 | Chevrier, B. / D'Orchymont, H. / Schalk, C. / Tarnus, C. / Moras, D. | ||||||
 引用 引用 | ジャーナル: Eur.J.Biochem. / 年: 1996 タイトル: The structure of the Aeromonas proteolytica aminopeptidase complexed with a hydroxamate inhibitor. Involvement in catalysis of Glu151 and two zinc ions of the co-catalytic unit. 著者: Chevrier, B. / D'Orchymont, H. / Schalk, C. / Tarnus, C. / Moras, D. #1: ジャーナル: Structure / 年: 1994タイトル: Crystal Structure of Aeromonas Proteolytica Aminopeptidase: A Prototypical Member of the Co-Catalytic Zinc Enzyme Family 著者: Chevrier, B. / Schalk, C. / D'Orchymont, H. / Rondeau, J.M. / Moras, D. / Tarnus, C. #2: ジャーナル: Arch.Biochem.Biophys. / 年: 1992タイトル: Rapid Purification of the Aeromonas Proteolytica Aminopeptidase: Crystallization and Preliminary X-Ray Data 著者: Schalk, C. / Remy, J.M. / Chevrier, B. / Moras, D. / Tarnus, C. | ||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 1igb.cif.gz | 92.3 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb1igb.ent.gz | 70.6 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 1igb.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| 文書・要旨 | 1igb_validation.pdf.gz | 702.7 KB | 表示 | wwPDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | 1igb_full_validation.pdf.gz | 708.6 KB | 表示 | |
| XML形式データ | 1igb_validation.xml.gz | 17.5 KB | 表示 | |
| CIF形式データ | 1igb_validation.cif.gz | 24.5 KB | 表示 | |
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/ig/1igbftp://data.pdbj.org/pub/pdb/validation_reports/ig/1igb | HTTPS FTP |
-関連構造データ










-リンク
 PDBj
PDBj
- 集合体
集合体
| 登録構造単位 | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 単位格子 |
|
-要素
| #1: タンパク質 | 分子量: 31427.350 Da / 分子数: 1 / 由来タイプ: 天然 / 由来: (天然) Vibrio proteolyticus (バクテリア) / 参照: UniProt: Q01693, bacterial leucyl aminopeptidase | ||||||
|---|---|---|---|---|---|---|---|
| #2: 化合物 |   分子量: 65.409 Da / 分子数: 2 / 由来タイプ: 合成 / 式: Zn 分子量: 65.409 Da / 分子数: 2 / 由来タイプ: 合成 / 式: Zn#3: 化合物 | ChemComp-IPO / | 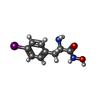  分子量: 306.100 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C9H11IN2O2 分子量: 306.100 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C9H11IN2O2#4: 水 | ChemComp-HOH / |  分子量: 18.015 Da / 分子数: 220 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 220 / 由来タイプ: 天然 / 式: H2OHas protein modification | Y | |
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.69 Å3/Da / 溶媒含有率: 54.27 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 結晶化 | *PLUS pH: 7 / 手法: 蒸気拡散法, ハンギングドロップ法 | ||||||||||||||||||||||||||||||||||||||||||
| 溶液の組成 | *PLUS
|
-データ収集
| 回折 | 平均測定温度: 291 K |
|---|---|
| 放射光源 | 由来: 回転陽極 / タイプ: SIEMENS / 波長: 1.5418 |
| 検出器 | タイプ: XENTRONICS / 検出器: AREA DETECTOR / 日付: 1993年9月1日 |
| 放射 | モノクロメーター: GRAPHITE(002) / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 波長: 1.5418 Å / 相対比: 1 |
| 反射 | 解像度: 2.3→50 Å / Num. obs: 15274 / % possible obs: 91.7 % / Observed criterion σ(I): 0 / 冗長度: 7.7 % / Rmerge(I) obs: 0.081 |
| 反射 | *PLUS Num. measured all: 118144 |
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: MOLECULAR REPLACEMENT SOFTWARE USED : X-PLOR STARTING MODEL FOR MOLECULAR REPLACEMENT: NATIVE PROTEIN (REFERENCE 1) 解像度: 2.3→8 Å / σ(F): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | Biso mean: 15.3 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.01 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 2.3→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ソフトウェア | *PLUS 名称: X-PLOR / バージョン: 3.1 / 分類: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化 | *PLUS Rfactor obs: 0.16 / Rfactor Rwork: 0.16 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 | *PLUS
|
