 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1gvo: STRUCTURE OF PENTAERYTHRITOL TETRANITRATE REDUCTASE AND COMPLEXED... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1gvo | ||||||
|---|---|---|---|---|---|---|---|
| Title | STRUCTURE OF PENTAERYTHRITOL TETRANITRATE REDUCTASE AND COMPLEXED WITH 2,4 DINITROPHENOL | ||||||
 Components Components | PENTAERYTHRITOL TETRANITRATE REDUCTASE | ||||||
 Keywords Keywords | OXIDOREDUCTASE / FLAVOENZYME / EXPLOSIVE DEGRADATION / STEROID BINDING | ||||||
| Function / homology |  Function and homology information Function and homology informationoxidoreductase activity, acting on the CH-CH group of donors, NAD or NADP as acceptor / FMN binding / cytosol Similarity search - Function | ||||||
| Biological species |  ENTEROBACTER CLOACAE (bacteria) ENTEROBACTER CLOACAE (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.38 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.38 Å | ||||||
 Authors Authors | Barna, T. / Moody, P.C.E. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2002 Title: Kinetic and Structural Basis of Reactivity of Pentaerythritol Tetranitrate Reductase with Nadph,2-Cyclohexenone Nitroesters and Nitroaromatic Explosives Authors: Khan, H. / Harris, R. / Barna, T. / Craig, D. / Bruce, N. / Munro, A. / Moody, P.C.E. / Scrutton, N. #1: Journal: J.Mol.Biol. / Year: 2001Title: Crystal Structure of Pentaerythritol Tetranitrate Reductase: "Flipped" Binding Geometries for Steroid Substrates in Different Redox States of the Enzyme Authors: Barna, T.M. / Khan, H. / Bruce, N.C. / Barsukov, I. / Scrutton, N.S. / Moody, P.C. #2: Journal: Acta Crystallogr.,Sect.D / Year: 1998 Title: Crystallization and Preliminary Diffraction Studies of Pentaerythritol Tetranitrate Reductase from Enterobacter Cloacae Pb2. Authors: Moody, P.C.E. / Shikotra, N. / French, C.E. / Bruce, N.C. / Scrutton, N.S. | ||||||
| History |
| ||||||
| Remark 700 | SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AB" IN EACH CHAIN ON SHEET RECORDS BELOW ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AB" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 8-STRANDED BARREL THIS IS REPRESENTED BY A 9-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1gvo.cif.gz | 103.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1gvo.ent.gz | 76.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1gvo.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gv/1gvoftp://data.pdbj.org/pub/pdb/validation_reports/gv/1gvo | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1gvqC  1gvrC  1gvsC  1h50S C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |















-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 39404.000 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: 2,4 DINITROPHENOL IS BOUND IN THE ACTIVE SITE / Source: (gene. exp.) ENTEROBACTER CLOACAE (bacteria) / Description: NCBI U68759. RECOMBINANT / Plasmid: PONR1 / Production host: |
|---|---|
| #2: Chemical | ChemComp-FMN / 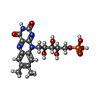  Mass: 456.344 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H21N4O9P Mass: 456.344 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H21N4O9P |
| #3: Chemical | ChemComp-DNF / 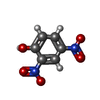  Mass: 184.106 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H4N2O5 Mass: 184.106 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H4N2O5 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 818 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 818 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.22 Å3/Da / Density % sol: 44.67 % | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6.2 / Details: pH 6.20 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, sitting drop / Details: Barna, T.M., (2001) J. Mol. Biol., 310, 433. / pH: 6.2 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SRS  / Beamline: PX9.6 / Wavelength: 0.87 / Beamline: PX9.6 / Wavelength: 0.87 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Jan 12, 1998 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.87 Å / Relative weight: 1 |
| Reflection | Resolution: 1.38→50 Å / Num. obs: 68596 / % possible obs: 96.2 % / Redundancy: 4.3 % / Biso Wilson estimate: 11.3 Å2 / Rmerge(I) obs: 0.038 / Net I/σ(I): 32.7 |
| Reflection shell | Resolution: 1.38→1.47 Å / % possible all: 79 |
| Reflection | *PLUS Num. obs: 69990 / % possible obs: 97.8 % / Num. measured all: 301258 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1H50 Resolution: 1.38→50 Å / Rfactor Rfree error: 0.003 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 12.7 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.38→50 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.38→1.47 Å / Rfactor Rfree error: 0.009 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor Rfree: 0.2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
