 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1h62: Structure of Pentaerythritol tetranitrate reductase in complex wi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1h62 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of Pentaerythritol tetranitrate reductase in complex with 1,4-androstadien-3,17-dione | ||||||
 Components Components | PENTAERYTHRITOL TETRANITRATE REDUCTASE | ||||||
 Keywords Keywords | OXIDOREDUCTASE / FLAVOENZYME / STEROID BINDING | ||||||
| Function / homology |  Function and homology information Function and homology informationoxidoreductase activity, acting on the CH-CH group of donors, NAD or NADP as acceptor / FMN binding / cytosol Similarity search - Function | ||||||
| Biological species |  ENTEROBACTER CLOACAE (bacteria) ENTEROBACTER CLOACAE (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.9 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | ||||||
 Authors Authors | Barna, T.M. / Moody, P.C.E. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2001 Title: Crystal Structure of Pentaerythritol Tetranitrate Reductase: "Flipped" Binding Geometries for Steroid Substrates in Different Redox States of the Enzyme Authors: Barna, T.M. / Khan, H. / Bruce, N.C. / Barsukov, I. / Scrutton, N.S. / Moody, P.C. | ||||||
| History |
| ||||||
| Remark 700 | SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AB" IN EACH CHAIN ON SHEET RECORDS BELOW ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AB" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 8-STRANDED BARREL THIS IS REPRESENTED BY A 9-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1h62.cif.gz | 95.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1h62.ent.gz | 70.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1h62.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/h6/1h62ftp://data.pdbj.org/pub/pdb/validation_reports/h6/1h62 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1h50C  1h51C  1h60C  1h61C  1h63C  1oyaS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 39404.000 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: NON-COVALENTLY BOUND FLAVIN MONONUCLEOTIDE / Source: (gene. exp.) ENTEROBACTER CLOACAE (bacteria) / Strain: PB2 / Plasmid: PONR1 / Production host: |
|---|---|
| #2: Chemical | ChemComp-ANB / 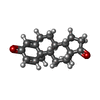  Mass: 284.393 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H24O2 / Comment: hormone*YM Mass: 284.393 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H24O2 / Comment: hormone*YM |
| #3: Chemical | ChemComp-FMN / 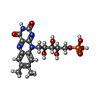  Mass: 456.344 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H21N4O9P Mass: 456.344 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H21N4O9P |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 484 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 484 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.3 Å3/Da / Density % sol: 38 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6.5 / Details: PEG 3000, 0.1M CACODYLIC ACID, 15% MEOH PH 6.8 | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, sitting drop / pH: 6.2 | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS IV IMAGE PLATE / Detector: IMAGE PLATE / Date: Jan 15, 2000 / Details: MIRRORS |
| Radiation | Monochromator: NI FILTERS / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→100 Å / Num. obs: 21069 / % possible obs: 94.4 % / Observed criterion σ(I): 0 / Redundancy: 2.5 % / Biso Wilson estimate: 19.9 Å2 / Rmerge(I) obs: 0.081 / Net I/σ(I): 9.5 |
| Reflection shell | Resolution: 1.9→1.97 Å / Rmerge(I) obs: 0.35 / Mean I/σ(I) obs: 5.5 / % possible all: 94.6 |
| Reflection | *PLUS Lowest resolution: 100 Å / Num. obs: 28269 / Num. measured all: 529105 / Rmerge(I) obs: 0.087 |
| Reflection shell | *PLUS Rmerge(I) obs: 0.382 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1OYA Resolution: 1.9→100 Å / Rfactor Rfree error: 0.007 / Data cutoff high absF: 1362424.18 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 24.6 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→100 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.9→2.02 Å / Rfactor Rfree error: 0.022 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 100 Å / Rfactor obs: 0.201 / Rfactor Rfree: 0.234 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
