 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1eh4: BINARY COMPLEX OF CASEIN KINASE-1 FROM S. POMBE WITH AN ATP COMPE... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1eh4 | ||||||
|---|---|---|---|---|---|---|---|
| Title | BINARY COMPLEX OF CASEIN KINASE-1 FROM S. POMBE WITH AN ATP COMPETITIVE INHIBITOR, IC261 | ||||||
 Components Components | CASEIN KINASE-1 | ||||||
 Keywords Keywords | TRANSFERASE / PROTEIN KINASE / CASEIN KINASE-1 / PROTEIN-INHIBITOR BINARY COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationfungal-type vacuole / regulation of endocytosis / endocytosis / protein tyrosine kinase activity / non-specific serine/threonine protein kinase / protein serine kinase activity / protein serine/threonine kinase activity / magnesium ion binding / signal transduction / ATP binding ...fungal-type vacuole / regulation of endocytosis / endocytosis / protein tyrosine kinase activity / non-specific serine/threonine protein kinase / protein serine kinase activity / protein serine/threonine kinase activity / magnesium ion binding / signal transduction / ATP binding / nucleus / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.8 Å X-RAY DIFFRACTION / Resolution: 2.8 Å | ||||||
 Authors Authors | Mashhoon, N. / Demaggio, A.J. / Tereshko, V. / Bergmeier, S.C. / Egli, M. / Hoekstra, M.F. / Kuret, J. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2000 Title: Crystal Structure of a Conformation-Selective Casein Kinase-1 Inhibitor Authors: Mashhoon, N. / Demaggio, A.J. / Tereshko, V. / Bergmeier, S.C. / Egli, M. / Hoekstra, M.F. / Kuret, J. #1: Journal: Embo J. / Year: 1995Title: Crystal Structure of Casein Kinase-1, a Phosphate-directed Protein Kinase Authors: Xu, R.M. / Carmel, G. / Sweet, R.M. / Kuret, J. / Cheng, X. #2: Journal: Proc.Natl.Acad.Sci.USA / Year: 1996Title: Structural Basis for Selectivity of the Isoquinoline Sulfonamide Family of Protein Kinase Inhibitors Authors: Xu, R.M. / Carmel, G. / Kuret, J. / Cheng, X. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1eh4.cif.gz | 131.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1eh4.ent.gz | 104.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1eh4.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/eh/1eh4ftp://data.pdbj.org/pub/pdb/validation_reports/eh/1eh4 | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |












-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | two molecules in the asymmetric unit are related by a 1/2c translation and a 4 degree rotation along crystal b axis |
-Components
| #1: Protein | Mass: 34377.238 Da / Num. of mol.: 2 / Fragment: CATALYTIC CORE RESIDUES 1 - 298 Source method: isolated from a genetically manipulated source Details: A 298 RESIDUE TRUNCATION MUTANT OF CKI1 Source: (gene. exp.) Description: SCHIZOSACCHAROMYCES POMBE / Plasmid: PT7B / Production host:  References: UniProt: P40233, Transferases; Transferring phosphorus-containing groups; Phosphotransferases with an alcohol group as acceptor #2: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 11 / Source method: obtained synthetically / Formula: SO4 / Details: IC261 was supplied by ICOS Mass: 96.063 Da / Num. of mol.: 11 / Source method: obtained synthetically / Formula: SO4 / Details: IC261 was supplied by ICOS#3: Chemical | 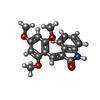  Mass: 311.332 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C18H17NO4 Mass: 311.332 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C18H17NO4#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 47 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 47 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.98 Å3/Da / Density % sol: 58.79 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 289 K / Method: vapor diffusion, hanging drop / pH: 4.2 Details: Ammonium Sulfate, Sodium Acetate, MPD , pH 4.2, VAPOR DIFFUSION, HANGING DROP, temperature 289K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 16 ℃ / pH: 7 / Method: vapor diffusion | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 133 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 / Wavelength: 1.5418 Å |
| Detector | Type: RIGAKU RAXIS IIC / Detector: IMAGE PLATE / Date: Mar 26, 1998 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.8→98 Å / Num. obs: 18625 / % possible obs: 95.5 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 1 / Redundancy: 5.3 % / Biso Wilson estimate: -0.2 Å2 / Rmerge(I) obs: 0.109 |
| Reflection shell | Resolution: 2.8→2.97 Å / Num. unique all: 2283 / % possible all: 76.4 |
| Reflection | *PLUS Lowest resolution: 98 Å / Num. measured all: 98304 |
| Reflection shell | *PLUS % possible obs: 76.4 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.8→20 Å / Rfactor Rfree error: 0.007 / Data cutoff high absF: 1991701.83 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 1 / Stereochemistry target values: standard cns values
| ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 31.71 Å2 / ksol: 0.304 e/Å3 | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 28 Å2
| ||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→20 Å
| ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.8→2.97 Å / Rfactor Rfree error: 0.024 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.5 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 0 / % reflection Rfree: 9.8 % | ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 28 Å2 | ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.374 / % reflection Rfree: 9.8 % / Rfactor Rwork: 0.328 |
