 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1ead: ATOMIC STRUCTURE OF THE CUBIC CORE OF THE PYRUVATE DEHYDROGENASE ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ead | ||||||
|---|---|---|---|---|---|---|---|
| Title | ATOMIC STRUCTURE OF THE CUBIC CORE OF THE PYRUVATE DEHYDROGENASE MULTIENZYME COMPLEX | ||||||
 Components Components | DIHYDROLIPOYL-TRANSACETYLASE | ||||||
 Keywords Keywords | DIHYDROLIPOAMIDE ACETYLTRANSFERASE | ||||||
| Function / homology |  Function and homology information Function and homology informationdihydrolipoyllysine-residue acetyltransferase / dihydrolipoyllysine-residue acetyltransferase activity / lipoic acid binding / pyruvate decarboxylation to acetyl-CoA / pyruvate dehydrogenase complex / cytoplasm Similarity search - Function | ||||||
| Biological species |  Azotobacter vinelandii (bacteria) Azotobacter vinelandii (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.6 Å X-RAY DIFFRACTION / Resolution: 2.6 Å | ||||||
 Authors Authors | Mattevi, A. / Hol, W.G.J. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1993 Title: Crystallographic analysis of substrate binding and catalysis in dihydrolipoyl transacetylase (E2p). Authors: Mattevi, A. / Obmolova, G. / Kalk, K.H. / Teplyakov, A. / Hol, W.G. #1: Journal: J.Mol.Biol. / Year: 1993Title: Three-Dimensional Structure of Lipoamide Dehydrogenase from Pseudomonas Fluorescens at 2.8 Angstroms Resolution. Analysis of Redox and Thermostability Properties Authors: Mattevi, A. / Obmolova, G. / Kalk, K.H. / Van Berkel, W.J. / Hol, W.G. #2: Journal: J.Mol.Biol. / Year: 1993Title: Refined Crystal Structure of the Catalytic Domain of Dihydrolipoyl Transacetylase (E2P) from Azotobacter Vinelandii at 2.6 Angstroms Resolution Authors: Mattevi, A. / Obmolova, G. / Kalk, K.H. / Westphal, A.H. / De Kok, A. / Hol, W.G. #3: Journal: Biochemistry / Year: 1993Title: Crystallographic Analysis of Substrate Binding and Catalysis in Dihydrolipoyl Transacetylase (E2P) Authors: Mattevi, A. / Obmolova, G. / Kalk, K.H. / Teplyakov, A. / Hol, W.G. | ||||||
| History |
| ||||||
| Remark 700 | SHEET SHEET A IS ACTUALLY A FOUR-STRANDED SHEET. IT CONTAINS TWO ADDITIONAL STRANDS A 3 ILE 409 PRO ...SHEET SHEET A IS ACTUALLY A FOUR-STRANDED SHEET. IT CONTAINS TWO ADDITIONAL STRANDS A 3 ILE 409 PRO 413 -1 A 4 THR 566 PHE 568 -1 FROM A THREEFOLD-RELATED SUBUNIT. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ead.cif.gz | 60.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ead.ent.gz | 45.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1ead.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ea/1eadftp://data.pdbj.org/pub/pdb/validation_reports/ea/1ead | HTTPS FTP |
|---|
-Related structure data















-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | x 24
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: RESIDUE 575 IS A CIS PROLINE. |
-Components
| #1: Protein | Mass: 26241.748 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Azotobacter vinelandii (bacteria)References: UniProt: P10802, dihydrolipoyllysine-residue acetyltransferase |
|---|---|
| #2: Chemical | ChemComp-CAO / 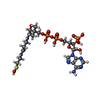  Mass: 783.534 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H36N7O17P3S Mass: 783.534 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H36N7O17P3S |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 45 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 45 / Source method: isolated from a natural source / Formula: H2O |
| Nonpolymer details | THESE COORDINATES ARE FOR THE BINARY COMPLEX WITH COA (RESIDUE 638). THE ELECTRON DENSITY SUGGESTS ...THESE COORDINATE |
| Sequence details | SEQUENCE ADVISORY NOTICE: DIFFERENCE BETWEEN SWISS-PROT AND PDB SEQUENCE. SWISS-PROT ENTRY NAME: ...SEQUENCE ADVISORY NOTICE: DIFFERENCE |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.55 Å3/Da / Density % sol: 72.97 % |
|---|---|
| Crystal grow | *PLUS pH: 7 / Method: unknown / Details: used macroseeding |
| Components of the solutions | *PLUS Conc.: 16 %sat / Common name: ammonium sulfate |
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 2.6 Å / Lowest resolution: 10 Å / Num. obs: 13596 / Rmerge(I) obs: 0.052 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.6→10 Å / Rfactor Rwork: 0.2 / Rfactor obs: 0.2 / σ(F): 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.6→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.6 Å / Lowest resolution: 10 Å / σ(F): 0 / Rfactor all: 0.2 / Rfactor Rwork: 0.2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
