 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1dvu: CRYSTAL STRUCTURE OF HUMAN TRANSTHYRETIN IN COMPLEX WITH DIBENZOF... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1dvu | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF HUMAN TRANSTHYRETIN IN COMPLEX WITH DIBENZOFURAN-4,6-DICARBOXYLIC ACID | ||||||
 Components Components | TRANSTHYRETIN | ||||||
 Keywords Keywords | HORMONE/GROWTH FACTOR / THYROXINE TRANSPORT / SIGNALING PROTEIN / HORMONE-GROWTH FACTOR COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationDefective visual phototransduction due to STRA6 loss of function / negative regulation of glomerular filtration / The canonical retinoid cycle in rods (twilight vision) / purine nucleobase metabolic process / hormone binding / Non-integrin membrane-ECM interactions / phototransduction, visible light / molecular sequestering activity / retinoid metabolic process / Retinoid metabolism and transport ...Defective visual phototransduction due to STRA6 loss of function / negative regulation of glomerular filtration / The canonical retinoid cycle in rods (twilight vision) / purine nucleobase metabolic process / hormone binding / Non-integrin membrane-ECM interactions / phototransduction, visible light / molecular sequestering activity / retinoid metabolic process / Retinoid metabolism and transport / hormone activity / azurophil granule lumen / Amyloid fiber formation / Neutrophil degranulation / protein-containing complex binding / protein-containing complex / : / extracellular exosome / extracellular region / identical protein binding Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 1.9 Å X-RAY DIFFRACTION / Resolution: 1.9 Å | ||||||
 Authors Authors | Klabunde, T. / Petrassi, H.M. / Oza, V.B. / Kelly, J.W. / Sacchettini, J.C. | ||||||
 Citation Citation | Journal: Nat.Struct.Biol. / Year: 2000 Title: Rational design of potent human transthyretin amyloid disease inhibitors. Authors: Klabunde, T. / Petrassi, H.M. / Oza, V.B. / Raman, P. / Kelly, J.W. / Sacchettini, J.C. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1dvu.cif.gz | 56.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1dvu.ent.gz | 41.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1dvu.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dv/1dvuftp://data.pdbj.org/pub/pdb/validation_reports/dv/1dvu | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1dvqC  1dvsC  1dvtC  1dvxC  1dvyC  1dvzC C: citing same article ( |
|---|---|
| Similar structure data |





















-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
| |||||||||
| Details | The biological assembly is a tetramer constructed from the homodimer (chain A and B) and a symmetry mate generated by the two-fold crystallographic axis. |
-Components
| #1: Protein | Mass: 13420.968 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Production host:  #2: Chemical | ChemComp-DBF / | 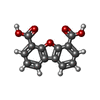  Mass: 256.210 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H8O5 Mass: 256.210 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H8O5#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 51 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 51 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.25 Å3/Da / Density % sol: 45.29 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 7.4 Details: potassium chloride, potassium phosphate, ammonium sulfate, pH 7.4, VAPOR DIFFUSION, HANGING DROP, temperature 293K | ||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: MACSCIENCE / Detector: IMAGE PLATE / Date: Oct 10, 1997 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.7→20 Å / Num. all: 26953 / Num. obs: 26953 / % possible obs: 98.4 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 5.83 % / Rmerge(I) obs: 0.075 / Net I/σ(I): 10.4 |
| Reflection shell | Resolution: 1.6→1.7 Å / Rmerge(I) obs: 0.316 / % possible all: 89.2 |
| Reflection | *PLUS Lowest resolution: 20 Å / Num. measured all: 157123 |
| Reflection shell | *PLUS % possible obs: 89.2 % |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.9→8 Å / σ(F): 2 / Stereochemistry target values: Engh & Huber Details: The structure contains two inhibitor molecules that bind in each of the two independent binding sites of the tetramer. Since the binding is along the 2-fold crystallographic axis, an ...Details: The structure contains two inhibitor molecules that bind in each of the two independent binding sites of the tetramer. Since the binding is along the 2-fold crystallographic axis, an occupancy of 0.5 corresponds to saturation of each of the binding sites.
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→8 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Classification: refinement | |||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.9 Å / Lowest resolution: 8 Å / σ(F): 2 / % reflection Rfree: 10 % / Rfactor obs: 0.198 / Rfactor Rfree: 0.22 | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS |
