 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1d8x | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF DNA SHEARED TANDEM G A BASE PAIRS | ||||||||||||||||||
 Components Components | 5'-D(* Keywords KeywordsDNA / TANDEM GA BASE PAIRS / GA MISMATCH | Function / homology | COBALT HEXAMMINE(III) / DNA |  Function and homology information Function and homology informationMethod |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.2 Å  Authors AuthorsGao, Y.-G. / Robinson, H. / Sanishvili, R. / Joachimiak, A. / Wang, A.H.-J. |  CitationJournal: Biochemistry / Year: 1999 CitationJournal: Biochemistry / Year: 1999Title: Structure and recognition of sheared tandem G x A base pairs associated with human centromere DNA sequence at atomic resolution. Authors: Gao, Y.G. / Robinson, H. / Sanishvili, R. / Joachimiak, A. / Wang, A.H. History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1d8x.cif.gz | 25.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1d8x.ent.gz | 15.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1d8x.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/d8/1d8xftp://data.pdbj.org/pub/pdb/validation_reports/d8/1d8x | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1d9rC  1dcrC C: citing same article ( |
|---|---|
| Similar structure data | |
| Other databases |
|





-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||||||||
| Unit cell |
| |||||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: DNA chain | Mass: 3094.042 Da / Num. of mol.: 2 / Source method: obtained synthetically #2: Chemical |   Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#3: Chemical | ChemComp-NCO / | 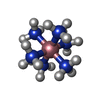  Mass: 161.116 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CoH18N6 Mass: 161.116 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CoH18N6#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 138 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 138 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.83 Å3/Da / Density % sol: 33 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion / pH: 7.5 Details: MPD, COBALT HEXAMMINE, MGCL2, TRIS, pH 7.5, VAPOR DIFFUSION, temperature 298K | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions |
| ||||||||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 123 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS IIC / Detector: IMAGE PLATE / Date: Feb 6, 1998 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.2→20 Å / Num. all: 12550 / Num. obs: 12550 / % possible obs: 81.7 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 1.6 % / Biso Wilson estimate: 31.9 Å2 / Rmerge(I) obs: 0.045 / Net I/σ(I): 16.5 |
| Reflection shell | Resolution: 1.2→1.24 Å / Redundancy: 1.25 % / Rmerge(I) obs: 0.39 / Mean I/σ(I) obs: 3.7 / % possible all: 46.8 |
| Reflection | *PLUS Observed criterion σ(F): 1 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.2→20 Å / Num. parameters: 4386 / Num. restraintsaints: 5330 / Cross valid method: A POSTERIORI / σ(F): 0 Stereochemistry target values: G.PARKINSON, J.VOJTECHOVSKY, L.CLOWNEY, A.T.BRUNGER, H.M.BERMAN
| |||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: SHELXL SWAT OPTION | |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 0 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.2→20 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL-97 / Classification: refinement | |||||||||||||||||||||||||||||||||
| Refinement | *PLUS Num. reflection obs: 11236 / σ(F): 0 / % reflection Rfree: 5 % / Rfactor obs: 0.182 / Rfactor Rwork: 0.188 | |||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
