 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1cd2: LIGAND INDUCED CONFORMATIONAL CHANGES IN THE CRYSTAL STRUCTURES O... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1cd2 | ||||||
|---|---|---|---|---|---|---|---|
| Title | LIGAND INDUCED CONFORMATIONAL CHANGES IN THE CRYSTAL STRUCTURES OF PNEUMOCYSTIS CARINII DIHYDROFOLATE REDUCTASE COMPLEXES WITH FOLATE AND NADP+ | ||||||
 Components Components | DIHYDROFOLATE REDUCTASE | ||||||
 Keywords Keywords | OXIDOREDUCTASE / ONE-CARBON METABOLISM | ||||||
| Function / homology |  Function and homology information Function and homology informationdihydrofolate metabolic process / dihydrofolate reductase / dihydrofolate reductase activity / folic acid metabolic process / tetrahydrofolate biosynthetic process / one-carbon metabolic process / NADP binding / mitochondrion Similarity search - Function | ||||||
| Biological species |  Pneumocystis carinii (fungus) Pneumocystis carinii (fungus) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.2 Å | ||||||
 Authors Authors | Cody, V. / Galitsky, N. / Luft, J.R. / Pangborn, W. / Blakley, R.L. / Gangjee, A. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1999 Title: Ligand-induced conformational changes in the crystal structures of Pneumocystis carinii dihydrofolate reductase complexes with folate and NADP+. Authors: Cody, V. / Galitsky, N. / Rak, D. / Luft, J.R. / Pangborn, W. / Queener, S.F. #1: Journal: Acta Crystallogr.,Sect.D / Year: 1997Title: Comparison of ternary complexes of Pneumocystis carinii and wild-type human dihydrofolate reductase with coenzyme NADPH and a novel classical antitumor furo[2,3-d]pyrimidine antifolate. Authors: Cody, V. / Galitsky, N. / Luft, J.R. / Pangborn, W. / Gangjee, A. / Devraj, R. / Queener, S.F. / Blakley, R.L. #2: Journal: J.Biol.Chem. / Year: 1995Title: Methotrexate-resistant variants of human dihydrofolate reductase with substitutions of leucine 22. Kinetics, crystallography, and potential as selectable markers. Authors: Lewis, W.S. / Cody, V. / Galitsky, N. / Luft, J.R. / Pangborn, W. / Chunduru, S.K. / Spencer, H.T. / Appleman, J.R. / Blakley, R.L. #3: Journal: J.Biol.Chem. / Year: 1994 Title: Methotrexate-resistant variants of human dihydrofolate reductase. Effects of Phe31 substitutions. Authors: Chunduru, S.K. / Cody, V. / Luft, J.R. / Pangborn, W. / Appleman, J.R. / Blakley, R.L. #4: Journal: Anti-Cancer Drug Des. / Year: 1992 Title: Crystal structure determination at 2.3 A of recombinant human dihydrofolate reductase ternary complex with NADPH and methotrexate-gamma-tetrazole. Authors: Cody, V. / Luft, J.R. / Ciszak, E. / Kalman, T.I. / Freisheim, J.H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1cd2.cif.gz | 60.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1cd2.ent.gz | 42.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1cd2.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cd/1cd2ftp://data.pdbj.org/pub/pdb/validation_reports/cd/1cd2 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1e26C  2cd2C  3cd2C  4cd2C  1dyrS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 23918.537 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: COMPLEXED WITH NADP+ AND FOLATE / Source: (gene. exp.) Pneumocystis carinii (fungus) / Plasmid: PT7-7 / Gene (production host): C-DNA P.CARINII DHFR / Production host:  |
|---|---|
| #2: Chemical | ChemComp-NAP /   Mass: 743.405 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H28N7O17P3 Mass: 743.405 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H28N7O17P3 |
| #3: Chemical | ChemComp-FOL / 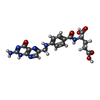  Mass: 441.397 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H19N7O6 Mass: 441.397 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H19N7O6 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 74 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 74 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.06 Å3/Da / Density % sol: 40.38 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6 Details: 10 ML PROTEIN, 1 MICROLITER 50MM MES, PH 6.5, 100MM KCL, 9 ML 50%(W/W)PEG 2K., pH 6.00 | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 6 / Method: unknown / Details: temperature gradient | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 287 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS II / Detector: IMAGE PLATE / Date: Aug 15, 1994 |
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.94→8 Å / Num. obs: 10428 / % possible obs: 84.7 % / Observed criterion σ(I): 2 / Redundancy: 3.09 % / Rmerge(I) obs: 0.086 |
| Reflection shell | Resolution: 2.18→2.2 Å / Redundancy: 3.6 % / Rmerge(I) obs: 0.052 / Mean I/σ(I) obs: 2.6 / Rsym value: 33.3 / % possible all: 90.3 |
| Reflection | *PLUS Highest resolution: 1.94 Å / Lowest resolution: 8 Å / % possible obs: 84.7 % / Observed criterion σ(I): 2 / Redundancy: 3.09 % |
| Reflection shell | *PLUS Highest resolution: 2.18 Å / Redundancy: 3.6 % / Mean I/σ(I) obs: 2.6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1DYR Resolution: 2.2→8 Å / σ(F): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.78 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: PROLSQ / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.2 Å / Lowest resolution: 8 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
