 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: EMDB / ID: EMD-7023 | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | Mitochondrial ATPase Protease YME1 | ||||||||||||||||||||||||
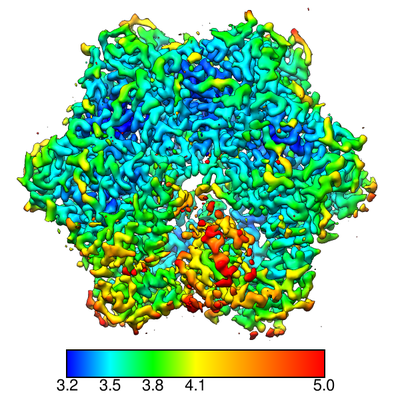
 マップデータ マップデータ | sharpened map | ||||||||||||||||||||||||
 試料 試料 |
| ||||||||||||||||||||||||
 キーワード キーワード | Mitochondrial / ATPase / Protease / HYDROLASE | ||||||||||||||||||||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報mitochondrial protein quality control / ATP-dependent peptidase activity / mitochondrion organization / metalloendopeptidase activity / mitochondrial inner membrane / ATP hydrolysis activity / ATP binding / metal ion binding 類似検索 - 分子機能 | ||||||||||||||||||||||||
| 生物種 |   | ||||||||||||||||||||||||
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 3.4 Å | ||||||||||||||||||||||||
 データ登録者 データ登録者 | Puchades C / Rampello AJ | ||||||||||||||||||||||||
| 資金援助 |  米国, 7件 米国, 7件
| ||||||||||||||||||||||||
 引用 引用 | ジャーナル: Science / 年: 2017 タイトル: Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing. 著者: Cristina Puchades / Anthony J Rampello / Mia Shin / Christopher J Giuliano / R Luke Wiseman / Steven E Glynn / Gabriel C Lander / 要旨: We present an atomic model of a substrate-bound inner mitochondrial membrane AAA+ quality control protease in yeast, YME1. Our ~3.4-angstrom cryo-electron microscopy structure reveals how the ...We present an atomic model of a substrate-bound inner mitochondrial membrane AAA+ quality control protease in yeast, YME1. Our ~3.4-angstrom cryo-electron microscopy structure reveals how the adenosine triphosphatases (ATPases) form a closed spiral staircase encircling an unfolded substrate, directing it toward the flat, symmetric protease ring. Three coexisting nucleotide states allosterically induce distinct positioning of tyrosines in the central channel, resulting in substrate engagement and translocation to the negatively charged proteolytic chamber. This tight coordination by a network of conserved residues defines a sequential, around-the-ring adenosine triphosphate hydrolysis cycle that results in stepwise substrate translocation. A hingelike linker accommodates the large-scale nucleotide-driven motions of the ATPase spiral relative to the planar proteolytic base. The translocation mechanism is likely conserved for other AAA+ ATPases. | ||||||||||||||||||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| ムービー |
ムービービューア |
|---|---|
| 構造ビューア | EMマップ: SurfViewMolmilJmol/JSmol |
| 添付画像 |
- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_7023.map.gz | 11.9 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-7023-v30.xmlemd-7023.xml | 33.6 KB 33.6 KB | 表示 表示 | EMDBヘッダ |
| FSC (解像度算出) | emd_7023_fsc.xml | 9 KB | 表示 | FSCデータファイル |
| 画像 | 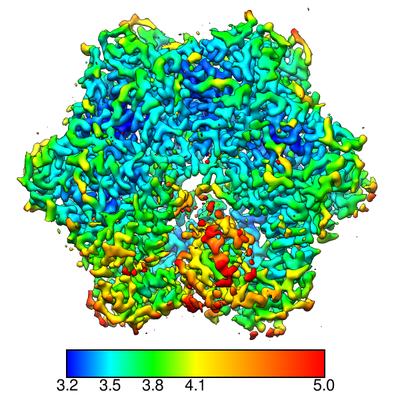 emd_7023.png emd_7023.png | 199.9 KB | ||
| Filedesc metadata | emd-7023.cif.gz | 7.1 KB | ||
| その他 | emd_7023_additional_1.map.gzemd_7023_additional_2.map.gzemd_7023_additional_3.map.gzemd_7023_half_map_1.map.gzemd_7023_half_map_2.map.gz | 1.7 MB 1.6 MB 4.4 MB 1.6 MB 1.6 MB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-7023ftp://ftp.pdbj.org/pub/emdb/structures/EMD-7023 http://ftp.pdbj.org/pub/emdb/structures/EMD-7023ftp://ftp.pdbj.org/pub/emdb/structures/EMD-7023 | HTTPS FTP |
-関連構造データ






-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| 「今月の分子」の関連する項目 |



-マップ
| ファイル | ダウンロード / ファイル: emd_7023.map.gz / 形式: CCP4 / 大きさ: 12.9 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | sharpened map | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 投影像・断面図 | 画像のコントロール
画像は Spider により作成 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 1.03 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 密度 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
CCP4マップ ヘッダ情報:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-添付データ
-追加マップ: focused refinement of step subunit, unsharpened
| ファイル | emd_7023_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | focused refinement of step subunit, unsharpened | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
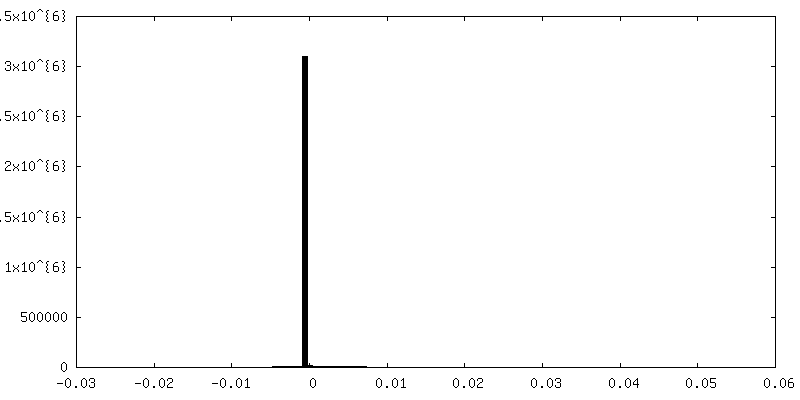
| 密度ヒストグラム |




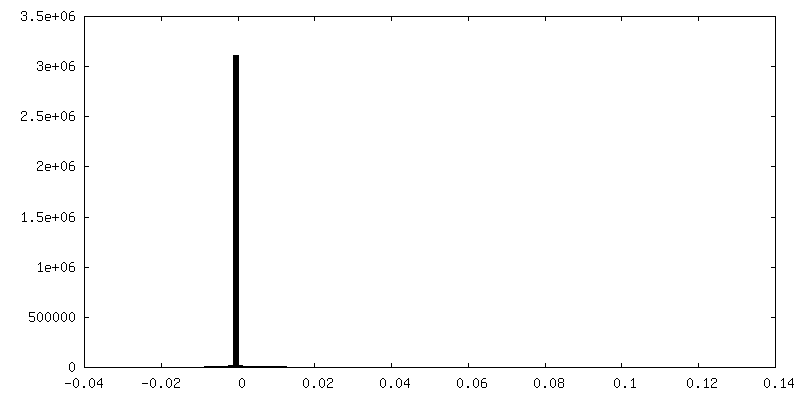
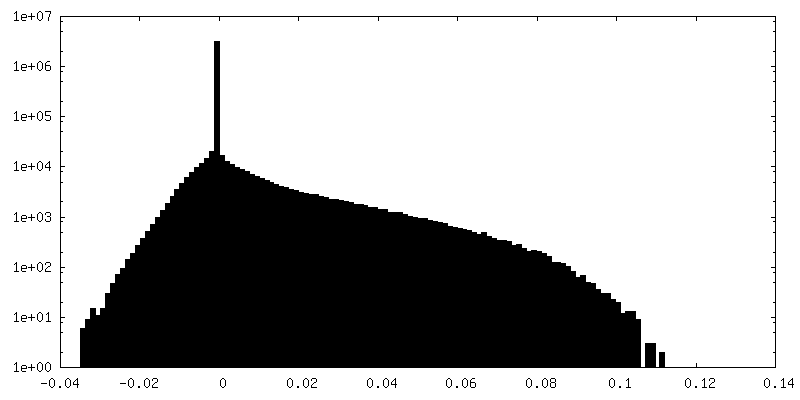
-追加マップ: focused refinement of step subunit, sharpened
| ファイル | emd_7023_additional_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | focused refinement of step subunit, sharpened | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
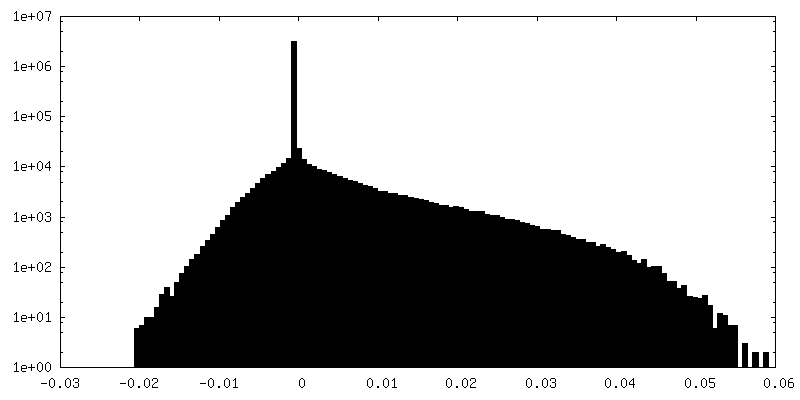
| 密度ヒストグラム |






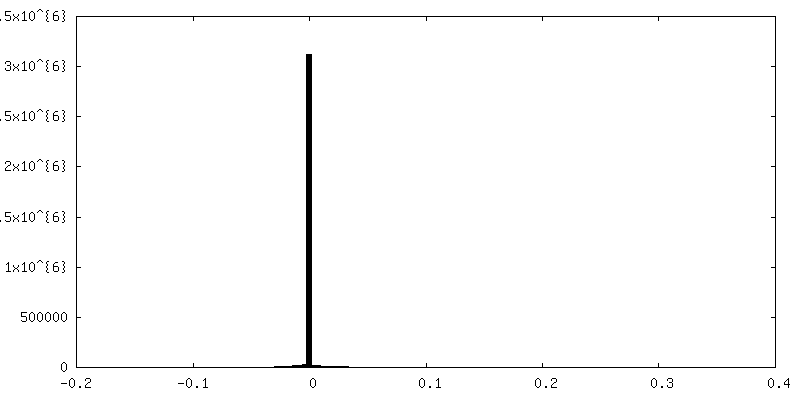
-追加マップ: unsharpened map
| ファイル | emd_7023_additional_3.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | unsharpened map | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |


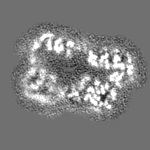
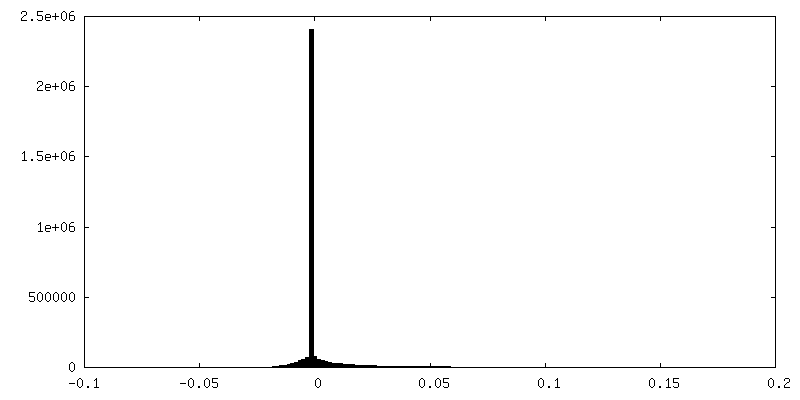
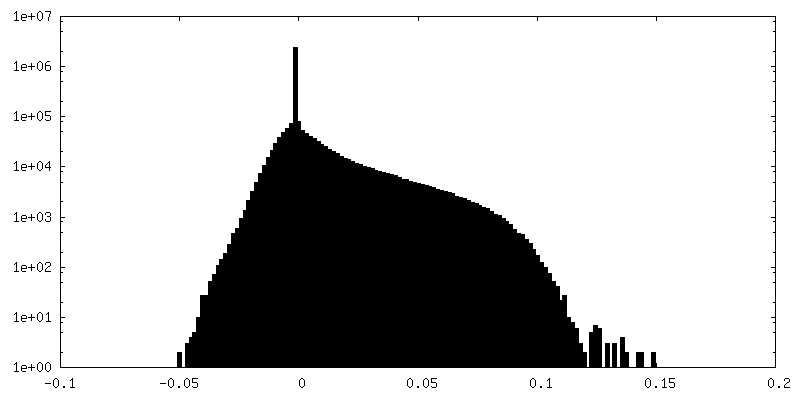
-ハーフマップ: focused refinement of step subunit, half map 2
| ファイル | emd_7023_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | focused refinement of step subunit, half map 2 | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |
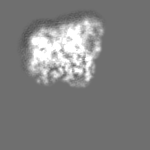





-ハーフマップ: focused refinement of step subunit, half map 1
| ファイル | emd_7023_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | focused refinement of step subunit, half map 1 | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |




- 試料の構成要素
試料の構成要素
-全体 : YME1 ATPase Protease Walker B mutant bound to substrate
| 全体 | 名称: YME1 ATPase Protease Walker B mutant bound to substrate |
|---|---|
| 要素 |
|
-超分子 #1: YME1 ATPase Protease Walker B mutant bound to substrate
| 超分子 | 名称: YME1 ATPase Protease Walker B mutant bound to substrate タイプ: complex / ID: 1 / 親要素: 0 / 含まれる分子: #1-#2 詳細: YME1 Walker B mutant was recombinantly expressed in E. coli, solved in presence of ATP with unknown bound substrate. |
|---|---|
| 由来(天然) | 生物種: |
| 分子量 | 理論値: 295 KDa |
-分子 #1: Mitochondrial inner membrane i-AAA protease supercomplex subunit YME1
| 分子 | 名称: Mitochondrial inner membrane i-AAA protease supercomplex subunit YME1 タイプ: protein_or_peptide / ID: 1 / 詳細: Walker B mutant / コピー数: 6 / 光学異性体: LEVO |
|---|---|
| 由来(天然) | 生物種: 株: RM11-1a |
| 分子量 | 理論値: 48.025758 KDa |
| 組換発現 | 生物種: |
| 配列 | 文字列: KFDDVCGCDE ARAELEEIVD FLKDPTKYES LGGKLPKGVL LTGPPGTGKT LLARATAGEA GVDFFFMSGS EFDEVYVGVG AKRIRDLFA QARSRAPAII FIDQLDAIGG KRNPKDQAYA KQTLNQLLVE LDGFSQTSGI IIIGATNFPE ALDKALTRPG R FDKVVNVD ...文字列: KFDDVCGCDE ARAELEEIVD FLKDPTKYES LGGKLPKGVL LTGPPGTGKT LLARATAGEA GVDFFFMSGS EFDEVYVGVG AKRIRDLFA QARSRAPAII FIDQLDAIGG KRNPKDQAYA KQTLNQLLVE LDGFSQTSGI IIIGATNFPE ALDKALTRPG R FDKVVNVD LPDVRGRADI LKHHMKKITL ADNVDPTIIA RGTPGLSGAE LANLVNQAAV YACQKNAVSV DMSHFEWAKD KI LMGAERK TMVLTDAARK ATAFHEAGHA IMAKYTNGAT PLYKATILPR GRALGITFQL PEMDKVDITK RECQARLDVC MGG KIAEEL IYGKDNTTSG CGSDLQSATG TARAMVTQYG MSDDVGPVNL SEEWESWSNK IRDIADNEVI ELLKDSEERA RRLL TKKNV ELHRLAQGLI EYETLDAHEI EQVCKGEKLA KLKT UniProtKB: AAA+ ATPase domain-containing protein |
-分子 #2: poly(UNK)
| 分子 | 名称: poly(UNK) / タイプ: protein_or_peptide / ID: 2 / 詳細: unknown polypeptide / コピー数: 1 / 光学異性体: LEVO |
|---|---|
| 由来(天然) | 生物種: |
| 分子量 | 理論値: 869.063 Da |
| 配列 | 文字列: (UNK)(UNK)(UNK)(UNK)(UNK)(UNK)(UNK)(UNK)(UNK)(UNK) |
-分子 #3: ADENOSINE-5'-TRIPHOSPHATE
| 分子 | 名称: ADENOSINE-5'-TRIPHOSPHATE / タイプ: ligand / ID: 3 / コピー数: 4 / 式: ATP |
|---|---|
| 分子量 | 理論値: 507.181 Da |
| Chemical component information |  ChemComp-ATP: |
-分子 #4: ZINC ION
| 分子 | 名称: ZINC ION / タイプ: ligand / ID: 4 / コピー数: 6 / 式: ZN |
|---|---|
| 分子量 | 理論値: 65.409 Da |
-分子 #5: MAGNESIUM ION
| 分子 | 名称: MAGNESIUM ION / タイプ: ligand / ID: 5 / コピー数: 4 / 式: MG |
|---|---|
| 分子量 | 理論値: 24.305 Da |
-分子 #6: ADENOSINE-5'-DIPHOSPHATE
| 分子 | 名称: ADENOSINE-5'-DIPHOSPHATE / タイプ: ligand / ID: 6 / コピー数: 1 / 式: ADP |
|---|---|
| 分子量 | 理論値: 427.201 Da |
| Chemical component information |  ChemComp-ADP: |
-実験情報
-構造解析
| 手法 | クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 緩衝液 | pH: 8 構成要素:
| |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| グリッド | モデル: Quantifoil R1.2/1.3 / 材質: GOLD / メッシュ: 300 / 前処理 - タイプ: PLASMA CLEANING / 前処理 - 時間: 6 sec. / 前処理 - 雰囲気: OTHER / 詳細: Gatan Solarus Plasma Cleaner | |||||||||||||||||||||
| 凍結 | 凍結剤: ETHANE / チャンバー内湿度: 100 % / チャンバー内温度: 298 K / 装置: FEI VITROBOT MARK IV |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TITAN KRIOS |
|---|---|
| 温度 | 最低: 70.0 K / 最高: 70.0 K |
| 詳細 | Data collected using Leginon |
| 撮影 | フィルム・検出器のモデル: GATAN K2 SUMMIT (4k x 4k) 検出モード: COUNTING / デジタル化 - サイズ - 横: 3710 pixel / デジタル化 - サイズ - 縦: 3838 pixel / デジタル化 - 画像ごとのフレーム数: 1-35 / 撮影したグリッド数: 3 / 実像数: 6098 / 平均露光時間: 7.0 sec. / 平均電子線量: 60.0 e/Å2 詳細: Images collected in counting mode at 5 frames per second. |
| 電子線 | 加速電圧: 300 kV / 電子線源:  FIELD EMISSION GUN FIELD EMISSION GUN |
| 電子光学系 | C2レンズ絞り径: 100.0 µm / 最大 デフォーカス(補正後): 2.5 µm / 最小 デフォーカス(補正後): 1.2 µm / 倍率(補正後): 51500 / 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELD / Cs: 2.7 mm / 最大 デフォーカス(公称値): 2.5 µm / 最小 デフォーカス(公称値): 1.2 µm / 倍率(公称値): 29000 |
| 試料ステージ | 試料ホルダーモデル: FEI TITAN KRIOS AUTOGRID HOLDER ホルダー冷却材: NITROGEN |
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |
+画像解析

-原子モデル構築 1
| 精密化 | 空間: REAL / プロトコル: AB INITIO MODEL / 温度因子: 100 |
|---|---|
| 得られたモデル |  PDB-6az0: |
