- EMDB-5645: Independent reconstruction of Mm-cpn cryo-EM density map from hal... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: EMDB / ID: EMD-5645
Title
Independent reconstruction of Mm-cpn cryo-EM density map from half dataset in the closed state (Training map)
Map data
Independent reconstruction of Mm-cpn cryo-EM density map from half dataset in the closed state (Training map)
Sample
Sample: Mm-cpn with 1mM ATP/AlFx
Protein or peptide: chaperonin
Keywords
modeling / independent reconstruction / cryo-EM model validation
Function / homology
Function and homology information
ATP-dependent protein folding chaperone / unfolded protein binding / ATP hydrolysis activity / ATP binding / identical protein binding / metal ion binding Similarity search - Function
Journal: Protein Sci / Year: 2013 Title: Cryo-EM model validation using independent map reconstructions. Authors: Frank DiMaio / Junjie Zhang / Wah Chiu / David Baker / Abstract: An increasing number of cryo-electron microscopy (cryo-EM) density maps are being generated with suitable resolution to trace the protein backbone and guide sidechain placement. Generating and ...An increasing number of cryo-electron microscopy (cryo-EM) density maps are being generated with suitable resolution to trace the protein backbone and guide sidechain placement. Generating and evaluating atomic models based on such maps would be greatly facilitated by independent validation metrics for assessing the fit of the models to the data. We describe such a metric based on the fit of atomic models with independent test maps from single particle reconstructions not used in model refinement. The metric provides a means to determine the proper balance between the fit to the density and model energy and stereochemistry during refinement, and is likely to be useful in determining values of model building and refinement metaparameters quite generally.
History
Deposition
May 1, 2013
-
Header (metadata) release
May 29, 2013
-
Map release
May 29, 2013
-
Update
Jun 12, 2013
-
Current status
Jun 12, 2013
Processing site: RCSB / Status: Released
-
Structure visualization
Movie
Surface view with section colored by density value
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Map data
Map data Sample
Sample Keywords
Keywords Function and homology information
Function and homology information Methanococcus maripaludis (archaea)
Methanococcus maripaludis (archaea) Authors
Authors Citation
Citation
 Structure visualization
Structure visualization
 Downloads & links
Downloads & links emd_5645_1.jpg
emd_5645_1.jpg http://ftp.pdbj.org/pub/emdb/structures/EMD-5645
http://ftp.pdbj.org/pub/emdb/structures/EMD-5645










 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)


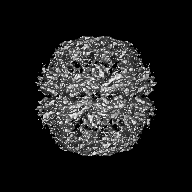






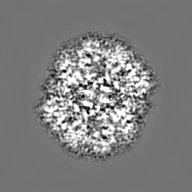










 Sample components
Sample components
 Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN