| Entry | Database: PDB / ID: 4cfl
|
|---|
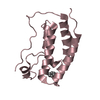
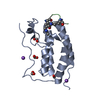
| Title | N-TERMINAL BROMODOMAIN OF HUMAN BRD4 WITH LY303511 |
|---|
 Components Components | BRD4 PROTEIN |
|---|
 Keywords Keywords | CELL CYCLE / INHIBITOR / HISTONE / EPIGENETIC READER / ANTAGONIST / KINASE |
|---|
| Function / homology |  Function and homology information Function and homology information
RNA polymerase II C-terminal domain binding / negative regulation of DNA damage checkpoint / P-TEFb complex binding / negative regulation by host of viral transcription / positive regulation of T-helper 17 cell lineage commitment / positive regulation of G2/M transition of mitotic cell cycle / histone reader activity / RNA polymerase II CTD heptapeptide repeat kinase activity / condensed nuclear chromosome / positive regulation of transcription elongation by RNA polymerase II ...RNA polymerase II C-terminal domain binding / negative regulation of DNA damage checkpoint / P-TEFb complex binding / negative regulation by host of viral transcription / positive regulation of T-helper 17 cell lineage commitment / positive regulation of G2/M transition of mitotic cell cycle / histone reader activity / RNA polymerase II CTD heptapeptide repeat kinase activity / condensed nuclear chromosome / positive regulation of transcription elongation by RNA polymerase II / transcription coregulator activity / lysine-acetylated histone binding / p53 binding / chromosome / chromatin organization / regulation of inflammatory response / positive regulation of canonical NF-kappaB signal transduction / Potential therapeutics for SARS / transcription coactivator activity / transcription cis-regulatory region binding / chromatin remodeling / DNA damage response / chromatin binding / regulation of transcription by RNA polymerase II / positive regulation of DNA-templated transcription / enzyme binding / positive regulation of transcription by RNA polymerase II / nucleoplasm / nucleusSimilarity search - Function Bromodomain protein 4, C-terminal / C-terminal domain of bromodomain protein 4 / NET domain superfamily / NET domain profile. / Brdt, bromodomain, repeat II / Brdt, bromodomain, repeat I / NET domain / Bromodomain extra-terminal - transcription regulation / Bromodomain-like / Histone Acetyltransferase; Chain A ...Bromodomain protein 4, C-terminal / C-terminal domain of bromodomain protein 4 / NET domain superfamily / NET domain profile. / Brdt, bromodomain, repeat II / Brdt, bromodomain, repeat I / NET domain / Bromodomain extra-terminal - transcription regulation / Bromodomain-like / Histone Acetyltransferase; Chain A / Bromodomain, conserved site / Bromodomain signature. / Bromodomain / Bromodomain profile. / bromo domain / Bromodomain / Bromodomain-like superfamily / Up-down Bundle / Mainly AlphaSimilarity search - Domain/homology |
|---|
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) |
|---|
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.32 Å |
|---|
 Authors Authors | Chung, C. / Dittmann, A. / Drewes, G. |
|---|
 Citation Citation | Journal: Acs Chem.Biol. / Year: 2014
Title: The Commonly Used Pi3-Kinase Probe Ly294002 is an Inhibitor of Bet Bromodomains.
Authors: Dittmann, A. / Werner, T. / Chung, C. / Savitski, M.M. / Falth Savitski, M. / Grandi, P. / Hopf, C. / Lindon, M. / Neubauer, G. / Prinjha, R.K. / Bantscheff, M. / Drewes, G. |
|---|
| History | | Deposition | Nov 18, 2013 | Deposition site: PDBE / Processing site: PDBE |
|---|
| Revision 1.0 | Jan 15, 2014 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Feb 26, 2014 | Group: Database references |
|---|
| Revision 1.2 | Mar 5, 2014 | Group: Database references |
|---|
|
|---|
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads


















 PDBj
PDBj
 Assembly
Assembly


 Mass: 306.358 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H18N2O2
Mass: 306.358 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H18N2O2
 Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2
 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3
Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 18.015 Da / Num. of mol.: 289 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 289 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: ID29 / Wavelength: 0.97625
/ Beamline: ID29 / Wavelength: 0.97625  Processing
Processing