Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 0.966 Å / Relative weight: 1
Reflection
Resolution: 1.44→38.359 Å / Num. obs: 71974 / % possible obs: 98.7 % / Redundancy: 3.1 % / CC1/2: 0.997 / Rmerge(I) obs: 0.075 / Net I/σ(I): 9.1
Reflection shell
Resolution: 1.44→1.469 Å / Redundancy: 3 % / Mean I/σ(I) obs: 2.1 / Num. unique obs: 3466 / CC1/2: 0.779 / Rrim(I) all: 0.431 / % possible all: 99.3
-
Processing
Software
Name
Version
Classification
REFMAC
5.8.0258
refinement
XDS
datareduction
Aimless
datascaling
Refinement
Method to determine structure: MOLECULAR REPLACEMENT Starting model: in house structure Resolution: 1.444→38.359 Å / Cor.coef. Fo:Fc: 0.957 / Cor.coef. Fo:Fc free: 0.94 / SU B: 1.324 / SU ML: 0.051 / Cross valid method: FREE R-VALUE / ESU R: 0.077 / ESU R Free: 0.076 Details: Hydrogens have been added in their riding positions
Rfactor
Num. reflection
% reflection
Rfree
0.21
1169
4.993 %
Rwork
0.1843
22242
-
all
0.186
-
-
obs
-
23411
98.677 %
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL PLUS MASK
Displacement parameters
Biso mean: 13.559 Å2
Baniso -1
Baniso -2
Baniso -3
1-
0.31 Å2
-0 Å2
0 Å2
2-
-
-0.217 Å2
-0 Å2
3-
-
-
-0.093 Å2
Refinement step
Cycle: LAST / Resolution: 1.444→38.359 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
1062
0
34
242
1338
Refine LS restraints
Refine-ID
Type
Dev ideal
Dev ideal target
Number
X-RAY DIFFRACTION
r_bond_refined_d
0.003
0.013
1130
X-RAY DIFFRACTION
r_bond_other_d
0.001
0.017
1033
X-RAY DIFFRACTION
r_angle_refined_deg
1.158
1.676
1535
X-RAY DIFFRACTION
r_angle_other_deg
1.207
1.594
2424
X-RAY DIFFRACTION
r_dihedral_angle_1_deg
6.131
5
128
X-RAY DIFFRACTION
r_dihedral_angle_2_deg
38.909
25.088
57
X-RAY DIFFRACTION
r_dihedral_angle_3_deg
10.846
15
198
X-RAY DIFFRACTION
r_dihedral_angle_4_deg
12.541
15
3
X-RAY DIFFRACTION
r_chiral_restr
0.058
0.2
143
X-RAY DIFFRACTION
r_gen_planes_refined
0.004
0.02
1273
X-RAY DIFFRACTION
r_gen_planes_other
0.001
0.02
208
X-RAY DIFFRACTION
r_nbd_refined
0.195
0.2
252
X-RAY DIFFRACTION
r_symmetry_nbd_other
0.164
0.2
937
X-RAY DIFFRACTION
r_nbtor_refined
0.169
0.2
531
X-RAY DIFFRACTION
r_symmetry_nbtor_other
0.081
0.2
396
X-RAY DIFFRACTION
r_xyhbond_nbd_refined
0.121
0.2
167
X-RAY DIFFRACTION
r_symmetry_nbd_refined
0.061
0.2
7
X-RAY DIFFRACTION
r_nbd_other
0.157
0.2
39
X-RAY DIFFRACTION
r_symmetry_xyhbond_nbd_refined
0.097
0.2
31
X-RAY DIFFRACTION
r_mcbond_it
0.814
1.614
509
X-RAY DIFFRACTION
r_mcbond_other
0.813
1.614
510
X-RAY DIFFRACTION
r_mcangle_it
1.41
3.611
636
X-RAY DIFFRACTION
r_mcangle_other
1.413
3.622
636
X-RAY DIFFRACTION
r_scbond_it
1.128
1.826
621
X-RAY DIFFRACTION
r_scbond_other
1.127
1.826
622
X-RAY DIFFRACTION
r_scangle_it
1.898
3.964
895
X-RAY DIFFRACTION
r_scangle_other
1.897
3.964
896
X-RAY DIFFRACTION
r_lrange_it
4.649
16.783
1411
X-RAY DIFFRACTION
r_lrange_other
4.196
15.568
1337
LS refinement shell
Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
Resolution (Å)
Rfactor Rfree
Num. reflection Rfree
Rfactor Rwork
Num. reflection Rwork
Rfactor all
Num. reflection all
Fsc free
Fsc work
% reflection obs (%)
WRfactor Rwork
1.444-1.481
0.293
82
0.246
1622
0.248
1719
0.833
0.868
99.1274
0.234
1.481-1.522
0.225
86
0.239
1573
0.238
1681
0.891
0.9
98.6913
0.223
1.522-1.566
0.233
73
0.224
1548
0.225
1644
0.91
0.916
98.601
0.211
1.566-1.614
0.211
77
0.214
1481
0.214
1572
0.926
0.926
99.1094
0.199
1.614-1.667
0.219
92
0.198
1444
0.199
1548
0.934
0.936
99.2248
0.18
1.667-1.725
0.253
66
0.202
1405
0.204
1479
0.912
0.936
99.4591
0.185
1.725-1.79
0.218
82
0.197
1358
0.198
1454
0.932
0.938
99.0371
0.182
1.79-1.863
0.213
72
0.18
1305
0.182
1392
0.92
0.947
98.9224
0.166
1.863-1.946
0.258
70
0.181
1248
0.185
1333
0.916
0.949
98.8747
0.169
1.946-2.041
0.165
67
0.169
1192
0.169
1282
0.965
0.963
98.2059
0.159
2.041-2.151
0.156
65
0.158
1133
0.158
1219
0.97
0.967
98.2773
0.15
2.151-2.281
0.193
61
0.161
1087
0.163
1158
0.95
0.961
99.1364
0.152
2.281-2.437
0.203
60
0.17
1034
0.172
1107
0.942
0.955
98.8257
0.163
2.437-2.632
0.213
58
0.185
945
0.187
1036
0.948
0.95
96.8147
0.179
2.632-2.882
0.246
39
0.176
875
0.178
946
0.939
0.954
96.6173
0.174
2.882-3.22
0.216
34
0.175
818
0.177
859
0.95
0.957
99.1851
0.178
3.22-3.713
0.148
27
0.16
742
0.16
775
0.969
0.973
99.2258
0.167
3.713-4.537
0.173
26
0.161
628
0.161
660
0.973
0.973
99.0909
0.171
4.537-6.372
0.35
18
0.212
505
0.217
533
0.922
0.952
98.1238
0.224
6.372-38.359
0.182
14
0.237
299
0.234
327
0.949
0.944
95.7187
0.265
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads



















 PDBj
PDBj
 Assembly
Assembly

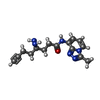
 Mass: 298.343 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H18N6O / Feature type: SUBJECT OF INVESTIGATION
Mass: 298.343 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H18N6O / Feature type: SUBJECT OF INVESTIGATION
 Mass: 62.068 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 18.015 Da / Num. of mol.: 242 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 242 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: ID30B / Wavelength: 0.966 Å
/ Beamline: ID30B / Wavelength: 0.966 Å Processing
Processing