 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3rnx | ||||||
|---|---|---|---|---|---|---|---|
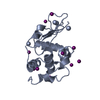
| Title | Crystal Structure of Lysozyme in 30% ethanol | ||||||
 Components Components | Lysozyme C | ||||||
 Keywords Keywords |  HYDROLASE / cytoplasmic vesicles HYDROLASE / cytoplasmic vesicles | ||||||
| Function / homology |  Function and homology informationAntimicrobial peptides / Neutrophil degranulation / beta-N-acetylglucosaminidase activity / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / killing of cells of another organism / defense response to Gram-negative bacterium / defense response to Gram-positive bacterium / defense response to bacterium ...Antimicrobial peptides / Neutrophil degranulation / beta-N-acetylglucosaminidase activity / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / killing of cells of another organism / defense response to Gram-negative bacterium / defense response to Gram-positive bacterium / defense response to bacterium / endoplasmic reticulum / extracellular space / identical protein binding / cytoplasm Function and homology informationAntimicrobial peptides / Neutrophil degranulation / beta-N-acetylglucosaminidase activity / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / killing of cells of another organism / defense response to Gram-negative bacterium / defense response to Gram-positive bacterium / defense response to bacterium ...Antimicrobial peptides / Neutrophil degranulation / beta-N-acetylglucosaminidase activity / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / killing of cells of another organism / defense response to Gram-negative bacterium / defense response to Gram-positive bacterium / defense response to bacterium / endoplasmic reticulum / extracellular space / identical protein binding / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Gallus gallus (chicken) Gallus gallus (chicken) | ||||||
| Method | X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.856 Å | ||||||
 Authors Authors | Sharma, P. / Solanki, A.K. / Ashish | ||||||
 Citation Citation | Journal: to be published Title: Crystal Structure of Lysozyme in 30% ethanol Authors: Sharma, P. / Solanki, A.K. / Ashish #1: Journal: Acta Crystallogr.,Sect.D / Year: 2005Title: Effect of alcohols on protein hydration: crystallographic analysis of hen egg-white lysozyme in the presence of alcohols Authors: Deshpande, A. / Nimsadkar, S. / Mande, S.C. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3rnx.cif.gz | 42.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3rnx.ent.gz | 27.6 KB | Display | PDB format |
| PDBx/mmJSON format | 3rnx.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/rn/3rnxftp://data.pdbj.org/pub/pdb/validation_reports/rn/3rnx | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1ykxS S: Starting model for refinement |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj









- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
| ||||||||||||
| Components on special symmetry positions |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 14331.160 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Gallus gallus (chicken) / References: UniProt: P00698, lysozyme |
|---|
-Non-polymers , 5 types, 106 molecules 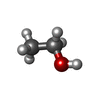




| #2: Chemical | ChemComp-EOH / Ethanol Mass: 46.068 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C2H6O Mass: 46.068 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C2H6O#3: Chemical | ChemComp-CL / Chloride Mass: 35.453 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: Cl#4: Chemical | ChemComp-ACT / | Acetate Mass: 59.044 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H3O2 Mass: 59.044 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H3O2#5: Chemical | ChemComp-NA / |  Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na#6: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 87 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.97 Å3/Da / Density % sol: 37.72 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 3.8 Details: 40 mM sodium acetate, 150mM sodium chloride, pH 3.8, VAPOR DIFFUSION, HANGING DROP, temperature 277K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-007 HF / Wavelength: 1.5418 Å |
| Detector | Type: MAR scanner 345 mm plate / Detector: IMAGE PLATE / Date: Oct 13, 2010 / Details: Mirrors |
| Radiation | Monochromator: mirrors / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.856→50 Å / Num. all: 10140 / Num. obs: 10140 / % possible obs: 99.5 % / Observed criterion σ(F): 2 / Observed criterion σ(I): 2 / Redundancy: 7.4 % / Biso Wilson estimate: 21.73 Å2 / Rmerge(I) obs: 0.04 / Net I/σ(I): 39.9 |
| Reflection shell | Resolution: 1.86→1.93 Å / Redundancy: 7.1 % / Rmerge(I) obs: 0.068 / Mean I/σ(I) obs: 33.6 / Num. unique all: 975 / % possible all: 99.9 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1ykx Resolution: 1.856→25.419 Å / Occupancy max: 1 / Occupancy min: 1 / FOM work R set: 0.865 / SU ML: 0.21 / Isotropic thermal model: Isotropic / σ(F): 0 / σ(I): 2 / Phase error: 19.26 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.83 Å / VDW probe radii: 1.1 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 58.026 Å2 / ksol: 0.476 e/Å3 | ||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 41.56 Å2 / Biso mean: 21.73 Å2 / Biso min: 10.34 Å2
| ||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.856→25.419 Å
| ||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 3
|
