 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3pvr: The Phenylacetyl-CoA monooxygenase PaaAC subcomplex with benzoyl-CoA -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3pvr | ||||||
|---|---|---|---|---|---|---|---|
| Title | The Phenylacetyl-CoA monooxygenase PaaAC subcomplex with benzoyl-CoA | ||||||
 Components Components |
| ||||||
 Keywords Keywords | OXIDOREDUCTASE / protein-protein complex / ferretin-like fold / bacterial multicomponent monooxygenase / structural genomics / Montreal-Kingston Bacterial Structural Genomics Initiative / BSGI | ||||||
| Function / homology |  Function and homology information Function and homology informationphenylacetyl-CoA 1,2-epoxidase / phenylacetyl-CoA 1,2-epoxidase complex / phenylacetyl-CoA 1,2-epoxidase activity / phenylacetate catabolic process / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.1 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.1 Å | ||||||
 Authors Authors | Cygler, M. / Grishin, A.M. / Montreal-Kingston Bacterial Structural Genomics Initiative (BSGI) | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2011 Title: Structural and Functional Studies of the Escherichia coli Phenylacetyl-CoA Monooxygenase Complex. Authors: Grishin, A.M. / Ajamian, E. / Tao, L. / Zhang, L. / Menard, R. / Cygler, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3pvr.cif.gz | 344.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3pvr.ent.gz | 280.4 KB | Display | PDB format |
| PDBx/mmJSON format | 3pvr.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/pv/3pvrftp://data.pdbj.org/pub/pdb/validation_reports/pv/3pvr | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3pvtC  3pvyC  3pw1C  3pw8C  3pwqC  1otkS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data | |
| Other databases |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 35788.535 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) References: UniProt: P76077, Oxidoreductases; Acting on paired donors, with incorporation or reduction of molecular oxygen; With NADH or NADPH as one donor, and incorporation of one atom of oxygen into the other donor | ||||||
|---|---|---|---|---|---|---|---|
| #2: Protein | Mass: 29109.629 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) References: UniProt: P76079, Oxidoreductases; Acting on paired donors, with incorporation or reduction of molecular oxygen; With NADH or NADPH as one donor, and incorporation of one atom of oxygen into the other donor #3: Chemical | ChemComp-BYC / | 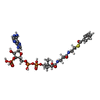  Mass: 871.640 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C28H40N7O17P3S Mass: 871.640 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C28H40N7O17P3S#4: Chemical | ChemComp-GOL /   Mass: 92.094 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C3H8O3#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 415 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 415 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.41 Å3/Da / Density % sol: 48.95 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: 100 mM PIPES, 5% PEG550 MME, 5% isopropanol, pH 6.5, VAPOR DIFFUSION, HANGING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 77.2 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CLSI  / Beamline: 08ID-1 / Wavelength: 0.97949 / Beamline: 08ID-1 / Wavelength: 0.97949 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: RAYONIX MX-300 / Detector: CCD / Date: Nov 25, 2009 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: DCM with cryo-cooled 1st crystal sagitally bent 2nd crystal followed by vertically focusing mirror Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97949 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.1→37.586 Å / Num. obs: 54874 / % possible obs: 99.5 % / Redundancy: 5.8 % / Rmerge(I) obs: 0.078 / Χ2: 0.905 / Net I/σ(I): 8.6 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1OTK Resolution: 2.1→37.586 Å / Cor.coef. Fo:Fc: 0.955 / Cor.coef. Fo:Fc free: 0.934 / WRfactor Rfree: 0.2053 / WRfactor Rwork: 0.1713 / Occupancy max: 1 / Occupancy min: 0.5 / FOM work R set: 0.8558 / SU B: 10.397 / SU ML: 0.123 / SU R Cruickshank DPI: 0.2093 / SU Rfree: 0.1734 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R Free: 0.173 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 125.63 Å2 / Biso mean: 36.6476 Å2 / Biso min: 14.99 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→37.586 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.1→2.154 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
