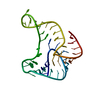
Rad6-Rad18 complex / positive regulation of chromosome segregation / Y-form DNA binding / nuclear inclusion body / DNA damage tolerance / polyubiquitin modification-dependent protein binding / protein monoubiquitination / protein autoubiquitination / replication fork / Recognition of DNA damage by PCNA-containing replication complex ...Rad6-Rad18 complex / positive regulation of chromosome segregation / Y-form DNA binding / nuclear inclusion body / DNA damage tolerance / polyubiquitin modification-dependent protein binding / protein monoubiquitination / protein autoubiquitination / replication fork / Recognition of DNA damage by PCNA-containing replication complex / RING-type E3 ubiquitin transferase / ubiquitin protein ligase activity / single-stranded DNA binding / E3 ubiquitin ligases ubiquitinate target proteins / site of double-strand break / damaged DNA binding / nuclear body / DNA repair / DNA damage response / ubiquitin protein ligase binding / centrosome / protein-containing complex binding / zinc ion binding / nucleoplasm / identical protein binding / nucleus / cytoplasm Similarity search - Function
E3 ubiquitin-protein ligase Rad18 / Rad18-like CCHC zinc finger / Zinc finger, C3HC4 type (RING finger) / SAP domain superfamily / SAP motif profile. / SAP domain / Putative DNA-binding (bihelical) motif predicted to be involved in chromosomal organisation / SAP domain / Rad18, zinc finger UBZ4-type / Zinc finger UBZ4-type profile. ...E3 ubiquitin-protein ligase Rad18 / Rad18-like CCHC zinc finger / Zinc finger, C3HC4 type (RING finger) / SAP domain superfamily / SAP motif profile. / SAP domain / Putative DNA-binding (bihelical) motif predicted to be involved in chromosomal organisation / SAP domain / Rad18, zinc finger UBZ4-type / Zinc finger UBZ4-type profile. / Zinc/RING finger domain, C3HC4 (zinc finger) / Herpes Virus-1 / Zinc finger, RING-type, conserved site / Zinc finger RING-type signature. / Ring finger / Zinc finger RING-type profile. / Zinc finger, RING-type / Zinc finger, RING/FYVE/PHD-type / 2-Layer Sandwich / Alpha Beta Similarity search - Domain/homology
Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 1.2828 Å / Relative weight: 1
Reflection
Resolution: 1.8→42.84 Å / Num. obs: 16560 / % possible obs: 99.9 % / Observed criterion σ(I): 3 / Redundancy: 17.3 % / Biso Wilson estimate: 18.4 Å2 / Rmerge(I) obs: 0.09 / Net I/σ(I): 24.3
Reflection shell
Resolution: 1.8→1.89 Å / Redundancy: 5.8 % / Rmerge(I) obs: 0.62 / Mean I/σ(I) obs: 2.5 / % possible all: 99.2
-
Processing
Software
Name
Version
Classification
XDS
datareduction
SCALA
datascaling
SHELX
phasing
PHASER
phasing
REFMAC
5.5.0099
refinement
Refinement
Method to determine structure: SAD Starting model: NONE Resolution: 1.8→42.86 Å / Cor.coef. Fo:Fc: 0.957 / Cor.coef. Fo:Fc free: 0.932 / SU B: 5.874 / SU ML: 0.082 / Cross valid method: THROUGHOUT / ESU R: 0.127 / ESU R Free: 0.127 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.22311
828
5 %
RANDOM
Rwork
0.17505
-
-
-
obs
0.1774
15732
99.92 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information HOMO SAPIENS (human)
HOMO SAPIENS (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads







 PDBj
PDBj






 Assembly
Assembly


 Mass: 65.409 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Zn Mass: 18.015 Da / Num. of mol.: 102 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 102 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: ID23-1 / Wavelength: 1.2828
/ Beamline: ID23-1 / Wavelength: 1.2828  Processing
Processing