 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2gve: Time-of-Flight Neutron Diffraction Structure of D-Xylose Isomerase -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2gve | ||||||
|---|---|---|---|---|---|---|---|
| Title | Time-of-Flight Neutron Diffraction Structure of D-Xylose Isomerase | ||||||
 Components Components | Xylose isomerase | ||||||
 Keywords Keywords | ISOMERASE / TIM barrel-beta-alpha-barrels / two metal binding sites / protonation states of residues | ||||||
| Function / homology |  Function and homology informationxylose isomerase / D-xylose metabolic process / xylose isomerase activity / magnesium ion binding / identical protein binding / cytoplasm Function and homology informationxylose isomerase / D-xylose metabolic process / xylose isomerase activity / magnesium ion binding / identical protein binding / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Streptomyces rubiginosus (bacteria) Streptomyces rubiginosus (bacteria) | ||||||
| Method | NEUTRON DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.2 Å | ||||||
 Authors Authors | Katz, A.K. / Li, X. / Carrell, H.L. / Hanson, B.L. / Langan, P. / Coates, L. / Schoenborn, B.P. / Glusker, J.P. / Bunick, G.J. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.Usa / Year: 2006 Title: Locating active-site hydrogen atoms in D-xylose isomerase: Time-of-flight neutron diffraction. Authors: Katz, A.K. / Li, X. / Carrell, H.L. / Hanson, B.L. / Langan, P. / Coates, L. / Schoenborn, B.P. / Glusker, J.P. / Bunick, G.J. #1: Journal: ACTA CRYSTALLOGR.,SECT.D / Year: 1994Title: Modes of binding substrates and their analogs to the enzyme D-xylose isomerase Authors: Carrell, H.L. / Hoier, H. / Glusker, J.P. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2gve.cif.gz | 188.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2gve.ent.gz | 153.1 KB | Display | PDB format |
| PDBx/mmJSON format | 2gve.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gv/2gveftp://data.pdbj.org/pub/pdb/validation_reports/gv/2gve | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2glkC  2gubC  1xibS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||
| Unit cell |
| |||||||||||||||
| Components on special symmetry positions |
| |||||||||||||||
| Details | homo-tetramer consisting of subunits related by crystallographic 222 symmetry |
-Components
| #1: Protein | Mass: 43283.297 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Streptomyces rubiginosus (bacteria) / References: UniProt: P24300, xylose isomerase | ||
|---|---|---|---|
| #2: Chemical |   Mass: 58.933 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Co Mass: 58.933 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Co#3: Chemical | ChemComp-DOD / | Heavy water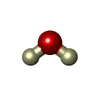  Mass: 18.015 Da / Num. of mol.: 512 / Source method: isolated from a natural source / Formula: D2O Mass: 18.015 Da / Num. of mol.: 512 / Source method: isolated from a natural source / Formula: D2O |
-Experimental details
-Experiment
| Experiment | Method: NEUTRON DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.78 Å3/Da / Density % sol: 55.75 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: liquid diffusion / pH: 8 Details: 50mM tris-HCl,38%AMSO4,2mM Mn2+ and 2mM Co2+,XI @ 125mg/ml, pH 8.0, LIQUID DIFFUSION, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 293 K | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Wavelength: 0.7-7.0 | |||||||||
| Detector | Type: Time-of-Flight Multiwire He3 neutron detector / Detector: AREA DETECTOR / Date: Sep 15, 2003 | |||||||||
| Radiation | Monochromator: chopper / Protocol: LAUE / Monochromatic (M) / Laue (L): L / Scattering type: neutron | |||||||||
| Radiation wavelength |
| |||||||||
| Reflection | Resolution: 1.8→20 Å / Num. all: 34394 / Num. obs: 34394 / % possible obs: 78 % / Observed criterion σ(F): 3 / Observed criterion σ(I): 1.5 / Rsym value: 0.185 | |||||||||
| Reflection shell | Resolution: 1.8→1.94 Å / Num. unique all: 3692 / Rsym value: 0.262 / % possible all: 39 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1XIB Resolution: 2.2→10 Å / σ(F): 3 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→10 Å
| ||||||||||||||||||||
| Refine LS restraints |
|
