 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1kv9: Structure at 1.9 A Resolution of a Quinohemoprotein Alcohol Dehyd... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1kv9 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure at 1.9 A Resolution of a Quinohemoprotein Alcohol Dehydrogenase from Pseudomonas putida HK5 | |||||||||
 Components Components | TYPE II QUINOHEMOPROTEIN ALCOHOL DEHYDROGENASE | |||||||||
 Keywords Keywords |  OXIDOREDUCTASE / Quinohemoprotein Alcohol Dehydrogenase / electron transfer OXIDOREDUCTASE / Quinohemoprotein Alcohol Dehydrogenase / electron transfer | |||||||||
| Function / homology |  Function and homology informationalcohol dehydrogenase (azurin) / pyrroloquinoline quinone binding / oxidoreductase activity, acting on CH-OH group of donors / outer membrane-bounded periplasmic space / electron transfer activity / oxidoreductase activity / calcium ion binding / heme binding / membrane / metal ion binding Function and homology informationalcohol dehydrogenase (azurin) / pyrroloquinoline quinone binding / oxidoreductase activity, acting on CH-OH group of donors / outer membrane-bounded periplasmic space / electron transfer activity / oxidoreductase activity / calcium ion binding / heme binding / membrane / metal ion bindingSimilarity search - Function | |||||||||
| Biological species |  Pseudomonas putida (bacteria) Pseudomonas putida (bacteria) | |||||||||
| Method | X-RAY DIFFRACTION / molecular replacement, MIR, MAD / Resolution: 1.9 Å | |||||||||
 Authors Authors | Chen, Z.-W. / Matsushita, K. / Yamashita, T. / Fujii, T. / Toyama, H. / Adachi, O. / Bellamy, H.D. / Mathews, F.S. | |||||||||
 Citation Citation | Journal: Structure / Year: 2002 Title: Structure at 1.9 A resolution of a quinohemoprotein alcohol dehydrogenase from Pseudomonas putida HK5. Authors: Chen, Z.W. / Matsushita, K. / Yamashita, T. / Fujii, T.A. / Toyama, H. / Adachi, O. / Bellamy, H.D. / Mathews, F.S. #1: Journal: J.Mol.Biol. / Year: 1996Title: Determination of the Gene Sequence and the Three-dimensional Structure at 2.4 Angstroms Resolution of Methanol Dehydrogenase from Methylophilus W3A1 Authors: Xia, Z. / Dai, W. / Zhang, Y. / White, S.A. / Boyd, G.D. / Mathews, F.S. | |||||||||
| History |
| |||||||||
| Remark 999 | sequence an appropriate sequence database reference was not available at the time of processing. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1kv9.cif.gz | 162.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1kv9.ent.gz | 122.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1kv9.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/kv/1kv9ftp://data.pdbj.org/pub/pdb/validation_reports/kv/1kv9 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4aahS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj








- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 72709.008 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Pseudomonas putida (bacteria) / References: UniProt: Q8GR64, EC: 1.1.99.- |
|---|
-Non-polymers , 7 types, 738 molecules 




| #2: Chemical | ChemComp-CA /  Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca |
|---|---|
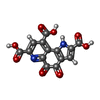
| #3: Chemical | ChemComp-PQQ / Pyrroloquinoline quinone Mass: 330.206 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H6N2O8 Mass: 330.206 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H6N2O8 |
| #4: Chemical | ChemComp-HEC / Heme C Mass: 618.503 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H34FeN4O4 Mass: 618.503 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H34FeN4O4 |
| #5: Chemical | ChemComp-EPE / HEPES Mass: 238.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM Mass: 238.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM |
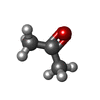
| #6: Chemical | ChemComp-ACN / Acetone Mass: 58.079 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H6O Mass: 58.079 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H6O |
| #7: Chemical | ChemComp-GOL / Glycerol Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 |
| #8: Water | ChemComp-HOH / WaterMass: 18.015 Da / Num. of mol.: 732 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.74 Å3/Da / Density % sol: 55 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: PEG 8000, sodium citrate, HEPES, 2-propanol, pH 7.5, VAPOR DIFFUSION, SITTING DROP, temperature 295.0K | ||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Details: used microseeding | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 Å |
| Detector | Type: RIGAKU RAXIS / Detector: IMAGE PLATE / Date: Jul 15, 1997 |
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→30 Å / Num. all: 48675 / Num. obs: 48672 / % possible obs: 87.1 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 3.5 % / Biso Wilson estimate: 12.8 Å2 / Rmerge(I) obs: 0.064 / Net I/σ(I): 18.2 |
| Reflection shell | Resolution: 1.9→1.97 Å / Redundancy: 2.8 % / Rmerge(I) obs: 0.215 / Mean I/σ(I) obs: 4.2 / Num. unique all: 2643 / % possible all: 47.3 |
| Reflection | *PLUS |
| Reflection shell | *PLUS % possible obs: 47.3 % |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: molecular replacement, MIR, MAD Starting model: PDB ENTRY 4AAH Resolution: 1.9→30 Å / Rfactor Rfree error: 0.003 / Isotropic thermal model: isotropic / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 21.6 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→30 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 30 Å / % reflection Rfree: 10 % | |||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| |||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rwork: 0.23 |
