 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1g7t: X-RAY STRUCTURE OF TRANSLATION INITIATION FACTOR IF2/EIF5B COMPLE... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1g7t | ||||||
|---|---|---|---|---|---|---|---|
| Title | X-RAY STRUCTURE OF TRANSLATION INITIATION FACTOR IF2/EIF5B COMPLEXED WITH GDPNP | ||||||
 Components Components | TRANSLATION INITIATION FACTOR IF2/EIF5B | ||||||
 Keywords Keywords | TRANSLATION / translational GTPase | ||||||
| Function / homology |  Function and homology information Function and homology informationtranslation initiation factor activity / GTPase activity / GTP binding / cytoplasm Similarity search - Function | ||||||
| Biological species |   Methanothermobacter thermautotrophicus (archaea) Methanothermobacter thermautotrophicus (archaea) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Roll-Mecak, A. / Cao, C. / Dever, T.E. / Burley, S.K. | ||||||
 Citation Citation | Journal: Cell(Cambridge,Mass.) / Year: 2000 Title: X-Ray structures of the universal translation initiation factor IF2/eIF5B: conformational changes on GDP and GTP binding. Authors: Roll-Mecak, A. / Cao, C. / Dever, T.E. / Burley, S.K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1g7t.cif.gz | 132 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1g7t.ent.gz | 99.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1g7t.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/g7/1g7tftp://data.pdbj.org/pub/pdb/validation_reports/g7/1g7t | HTTPS FTP |
|---|
-Related structure data










-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 66340.391 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Methanothermobacter thermautotrophicus (archaea)Production host:  |
|---|---|
| #2: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg |
| #3: Chemical | ChemComp-GNP / 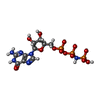  Mass: 522.196 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N6O13P3 Mass: 522.196 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N6O13P3Comment: GppNHp, GMPPNP, energy-carrying molecule analogue*YM |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 328 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 328 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.53 Å3/Da / Density % sol: 65.12 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 5.6 Details: 0.1M Cacodylate pH 5.6, 18% PEG4000, 0.2M Lithium Sulfate, VAPOR DIFFUSION, HANGING DROP, temperature 293K | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 5.8 | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 95 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X25 / Wavelength: 0.95 Å / Beamline: X25 / Wavelength: 0.95 Å |
| Detector | Type: BRANDEIS / Detector: CCD / Date: Apr 28, 2000 |
| Radiation | Monochromator: graphite / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.95 Å / Relative weight: 1 |
| Reflection | Resolution: 2→22 Å / Num. all: 61231 / Num. obs: 58931 / % possible obs: 97.2 % / Observed criterion σ(F): 2 / Observed criterion σ(I): 2 / Redundancy: 6 % / Rmerge(I) obs: 0.066 / Net I/σ(I): 17 |
| Reflection shell | Resolution: 2→2.05 Å / Redundancy: 6 % / Rmerge(I) obs: 0.17 / % possible all: 86 |
| Reflection shell | *PLUS % possible obs: 86 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2→22 Å / σ(F): 2 / σ(I): 2 / Stereochemistry target values: Engh & Huber Details: In REMARK 500, the covalent bonds which deviate from the dictionary values by more than 6RMSD are from residues for which the electron density is very poor beyond (and including) the CB atom.
| ||||||||||||||||||||
| Displacement parameters |
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→22 Å
| ||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||
| Software | *PLUS Name: CNS / Classification: refinement | ||||||||||||||||||||
| Refine LS restraints | *PLUS
|
